

ADAP is required for normal alphaIIbbeta3 activation by VWF/GP Ib-IX-V and other agonists
- PMID: 17003372
- PMCID: PMC1785130
- DOI: 10.1182/blood-2006-05-022301
ADAP is required for normal alphaIIbbeta3 activation by VWF/GP Ib-IX-V and other agonists
Abstract
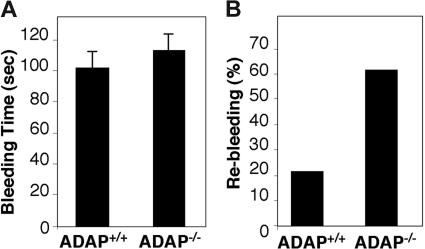
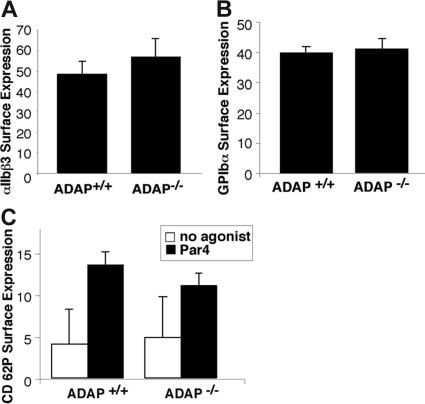
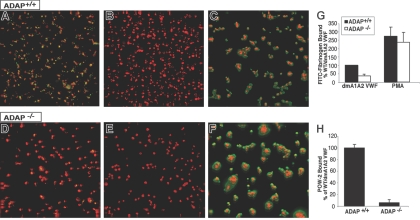
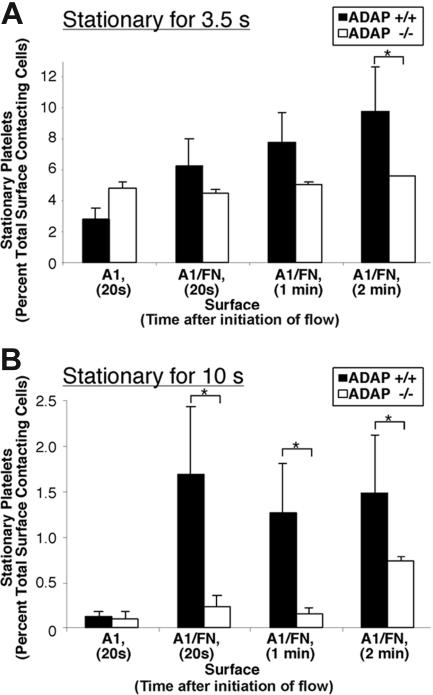
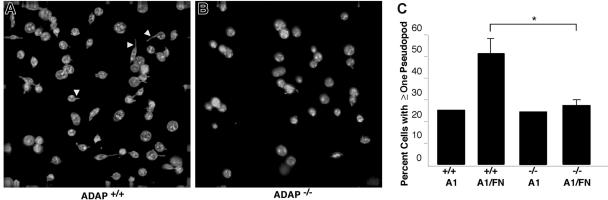
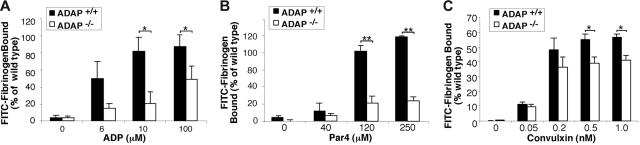
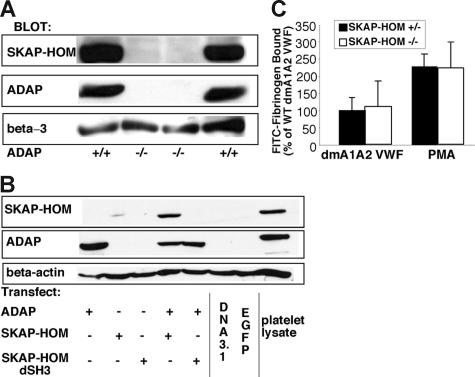
Interaction between von Willebrand factor (VWF) and platelet GP Ib-IX-V is required for hemostasis, in part because intracellular signals from VWF/GP Ib-IX-V activate the ligand-binding function of integrin alphaIIbbeta3. Because they also induce tyrosine phosphorylation of the ADAP adapter, we investigated ADAP's role in GP Ib-IX-V signal transduction. Fibrinogen or ligand-mimetic POW-2 Fab binding to alphaIIbbeta3 was stimulated by adhesion of ADAP+/+ murine platelets to dimeric VWF A1A2 but was significantly reduced in ADAP-/- platelets (P<.01). alphaIIbbeta3 activation by ADP or a Par4 thrombin receptor agonist was also decreased in ADAP-/- platelets. ADAP stabilized the expression of another adapter, SKAP-HOM, via interaction with the latter's SH3 domain. However, no abnormalities in alphaIIbbeta3 activation were observed in SKAP-HOM-/- platelets, which express normal ADAP levels, further implicating ADAP as a modulator of alphaIIbbeta3 function. Under shear flow conditions over a combined surface of VWF A1A2 and fibronectin to test interactions involving GP Ib-IX-V and alphaIIbbeta3, respectively, ADAP-/- platelets displayed reduced alphaIIbbeta3-dependent stable adhesion. Furthermore, ADAP-/- mice demonstrated increased rebleeding from tail wounds. These studies establish ADAP as a component of inside-out signaling pathways that couple GP Ib-IX-V and other platelet agonist receptors to alphaIIbbeta3 activation.
Figures
References
-
- Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289–297. - PubMed
-
- Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657–666. - PubMed
-
- Sakariassen KS, Bolhuis PA, Sixma JJ. Human blood platelet adhesion to artery subendothelium is mediated by factor VIII-Von Willebrand factor bound to the subendothelium. Nature. 1979;279:636–638. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
