

EBNA-3B- and EBNA-3C-regulated cellular genes in Epstein-Barr virus-immortalized lymphoblastoid cell lines
- PMID: 17005691
- PMCID: PMC1617319
- DOI: 10.1128/JVI.00854-06
EBNA-3B- and EBNA-3C-regulated cellular genes in Epstein-Barr virus-immortalized lymphoblastoid cell lines
Abstract
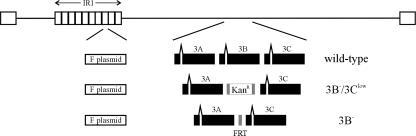
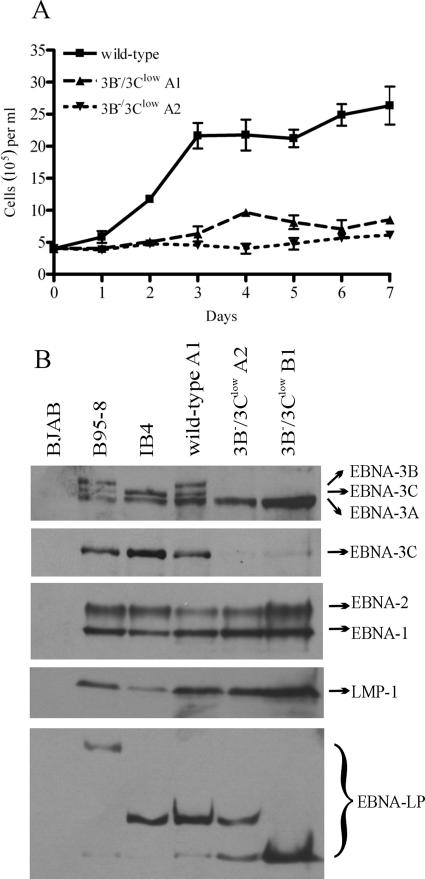
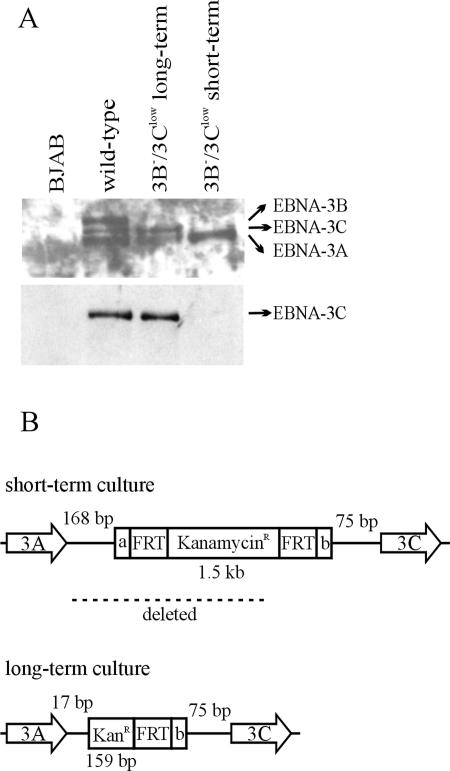
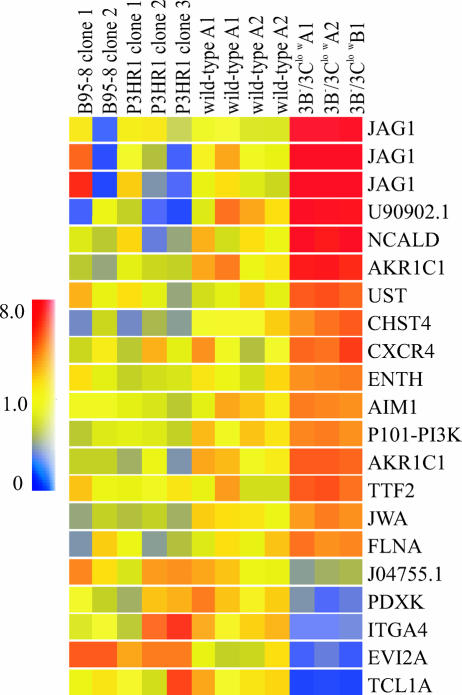
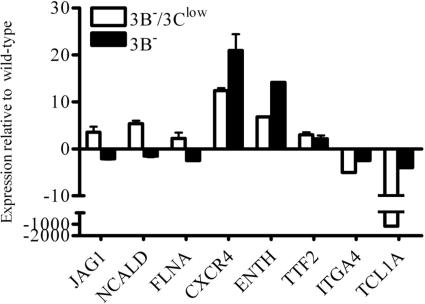
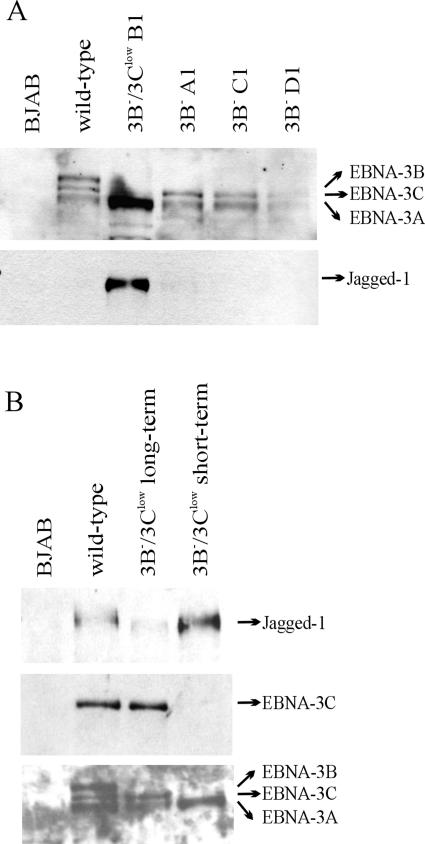
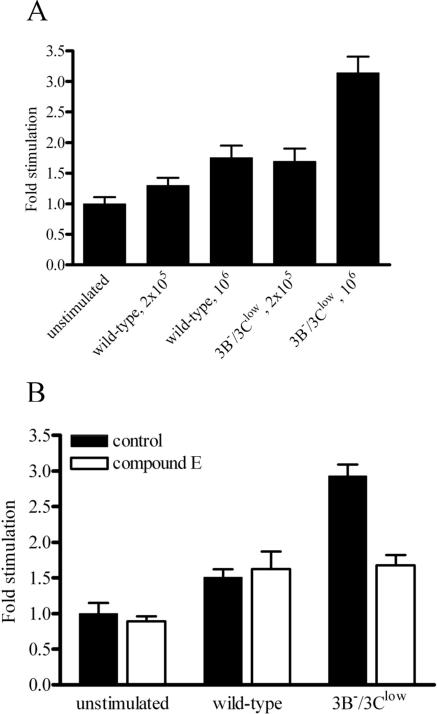
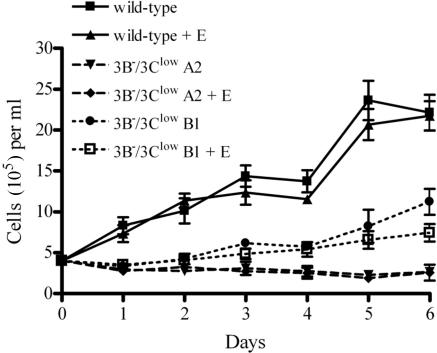
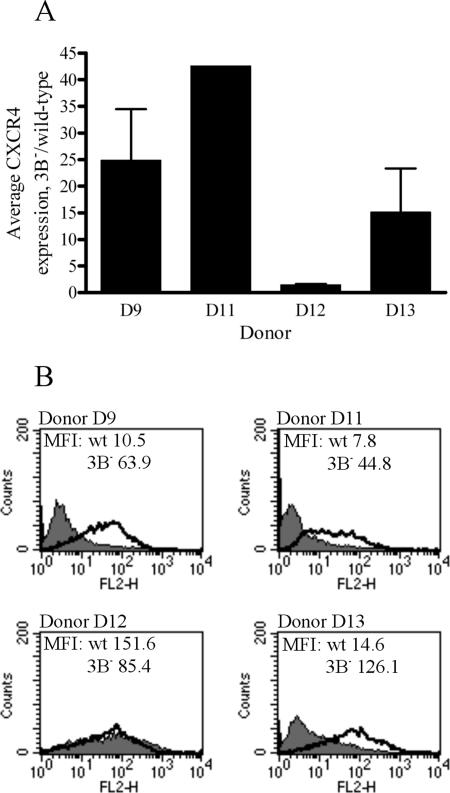
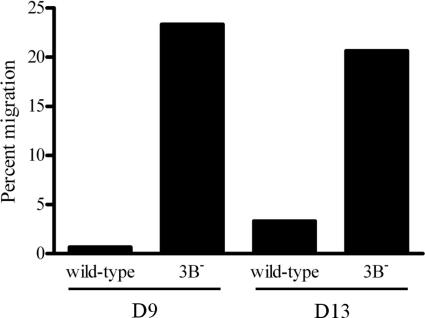
The cellular pathways that Epstein-Barr virus (EBV) manipulates in order to effect its lifelong persistence within hosts and facilitate its transmission between hosts are not well understood. The EBV nuclear antigen 3 (EBNA-3) family of latent infection proteins consists of transcriptional regulators that influence viral and cellular gene expression in EBV-infected cells. To identify EBNA-3B- and EBNA-3C-regulated cellular genes potentially important for virus infection in vivo, we studied a lymphoblastoid cell line (LCL) infected with an unusual EBV mutant, where a genetic manipulation to delete EBNA-3B also resulted in a significant decrease in EBNA-3C expression and slower than normal growth (3B(-)/3C(low)). Transcriptional profiling was performed on the 3B(-)/3C(low) LCLs, and comparison of mutant and wild-type LCL profiles resulted in a group of 21 probe sets representing 16 individual genes showing statistically significant differences in expression. Further quantitative reverse transcription-PCR analyses comparing 3B(-)/3C(low) LCLs to a previously described EBNA-3B mutant (3B(-)) where EBNA-3C expression was normal revealed three potential EBNA-3B-repressed genes, three potential EBNA-3C-repressed genes, and two potential EBNA-3C-activated genes. The most highly EBNA-3C-repressed gene was Jagged1, a cell surface ligand and inducer of the Notch receptor signaling pathway that is usurped by EBV genes essential for B-cell immortalization. 3B(-)/3C(low) LCLs expressed increased levels of Jagged1 protein and were able to more efficiently induce functional Notch signaling, and this signaling was dependent on Notch cleavage by gamma-secretase. However, inhibiting gamma-secretase-mediated Notch cleavage did not rescue 3B(-)/3C(low) LCL growth, suggesting that EBNA-3C-mediated repression of this signaling pathway did not contribute to LCL growth in tissue culture. Similarly, expression of the chemokine receptor CXCR4 was reproducibly upregulated in EBNA-3B-null LCLs. Since deletion of EBNA-3B has no significant impact on B-cell immortalization in tissue culture, this finding suggested that EBNA-3B-mediated regulation of CXCR4 may be an important viral strategy for alteration of B-cell homing in the infected host. These studies identify two cellular genes that do not contribute to EBV-induced B-cell growth but whose expression levels are strongly EBNA-3 regulated in EBV-infected primary B cells. These EBV-manipulated cellular pathways may be important for virus survival or transmission in humans, and their independence from EBV-induced B-cell growth makes them potential targets for testing in vivo with the rhesus lymphocryptovirus animal model for EBV infection.
Figures
References
-
- Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395-404. - PubMed
-
- D'Apuzzo, M., A. Rolink, M. Loetscher, J. A. Hoxie, I. Clark-Lewis, F. Melchers, M. Baggiolini, and B. Moser. 1997. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 27:1788-1793. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
