

Molecular system bioenergetics: regulation of substrate supply in response to heart energy demands
- PMID: 17008367
- PMCID: PMC1890373
- DOI: 10.1113/jphysiol.2006.120584
Molecular system bioenergetics: regulation of substrate supply in response to heart energy demands
Abstract
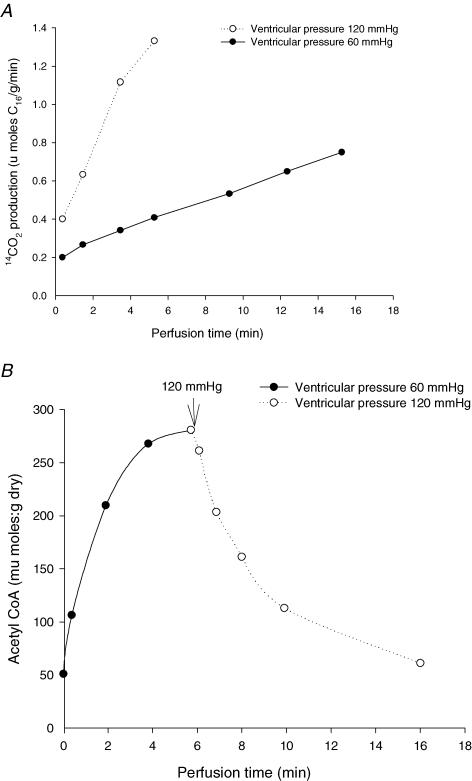
This review re-evaluates regulatory aspects of substrate supply in heart. In aerobic heart, the preferred substrates are always free fatty acids, and workload-induced increase in their oxidation is observed at unchanged global levels of ATP, phosphocreatine and AMP. Here, we evaluate the mechanisms of regulation of substrate supply for mitochondrial respiration in muscle cells, and show that a system approach is useful also for revealing mechanisms of feedback signalling within the network of substrate oxidation and particularly for explaining the role of malonyl-CoA in regulation of fatty acid oxidation in cardiac muscle. This approach shows that a key regulator of fatty acid oxidation is the energy demand. Alterations in malonyl-CoA would not be the reason for, but rather the consequence of, the increased fatty acid oxidation at elevated workloads, when the level of acetyl-CoA decreases due to shifts in the kinetics of the Krebs cycle. This would make malonyl-CoA a feedback regulator that allows acyl-CoA entry into mitochondrial matrix space only when it is needed. Regulation of malonyl-CoA levels by AMPK does not seem to work as a master on-off switch, but rather as a modulator of fatty acid import.
Figures

References
-
- Assifi MM, Suchankova G, Constant S, Prentki M, Saha AK, Ruderman NB. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am J Physiol Endocrinol Metab. 2005;289:E794–E800. - PubMed
-
- Balaban RS, Heineman FW. Interaction of oxidative phosphorylation and work in the heart in vivo. News Physiol Sci. 1989;4:215–218.
-
- Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolites in the in vivo paced mammalian heart. Science. 1986;232:1121–1123. - PubMed
-
- Beauloye C, Marsin AS, Bertrand L, Vanoverschelde JL, Rider MH, Hue L. The stimulation of heart glycolysis by increased workload does not require AMP activated protein kinase but a wortmannin-sensitive mechanism. FEBS Lett. 2002;531:324–328. - PubMed
-
- Bing RJ. Cardiac metabolism. Physiol Rev. 1965;45:171–213. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources