

Death receptor ligation triggers membrane scrambling between Golgi and mitochondria
- PMID: 17008914
- PMCID: PMC2941902
- DOI: 10.1038/sj.cdd.4402043
Death receptor ligation triggers membrane scrambling between Golgi and mitochondria
Abstract
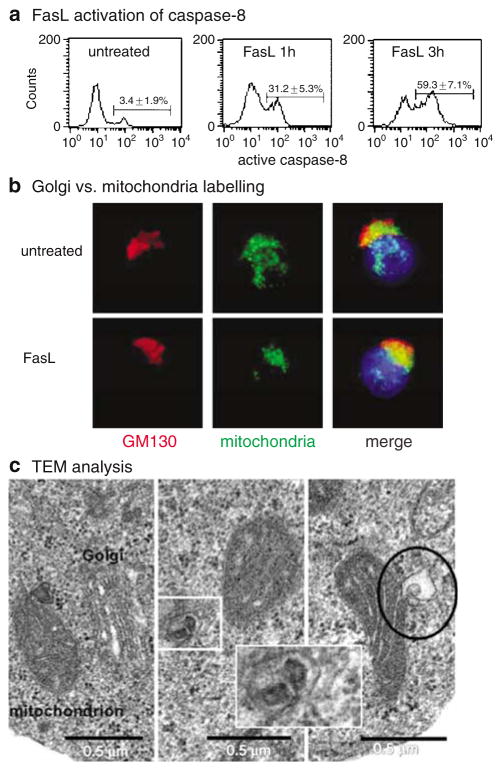
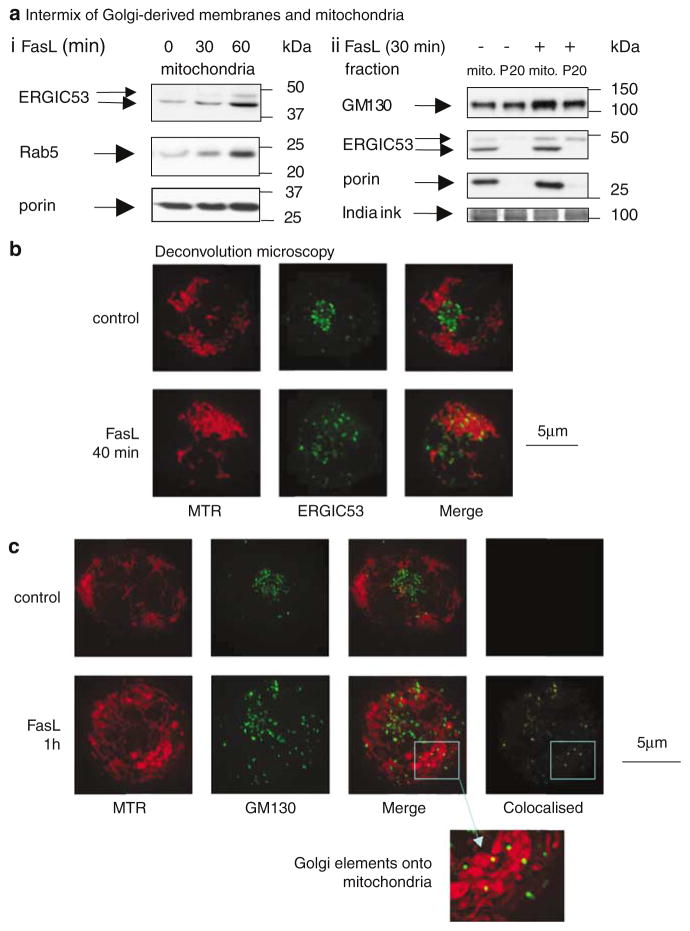
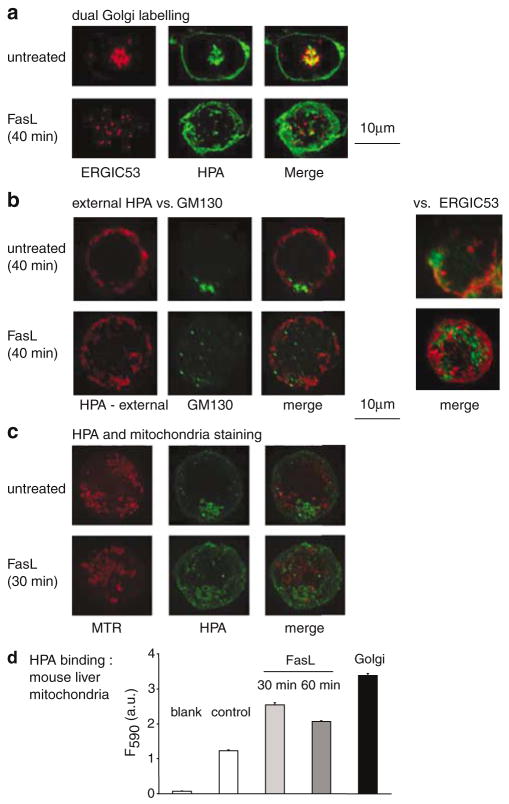
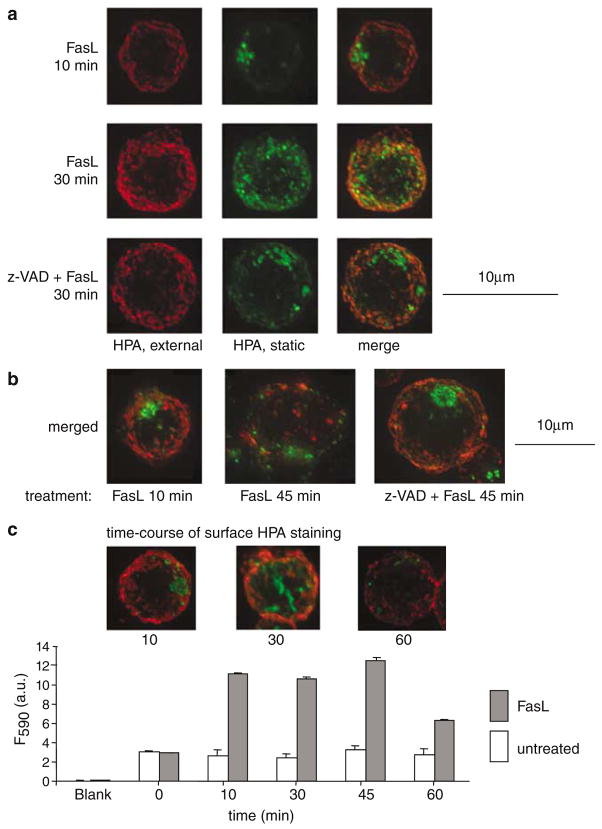
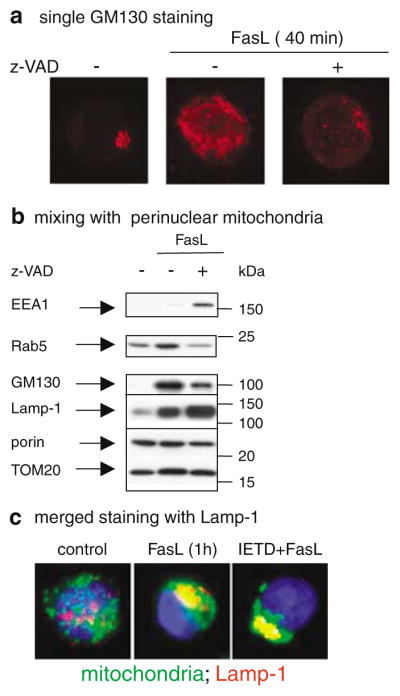
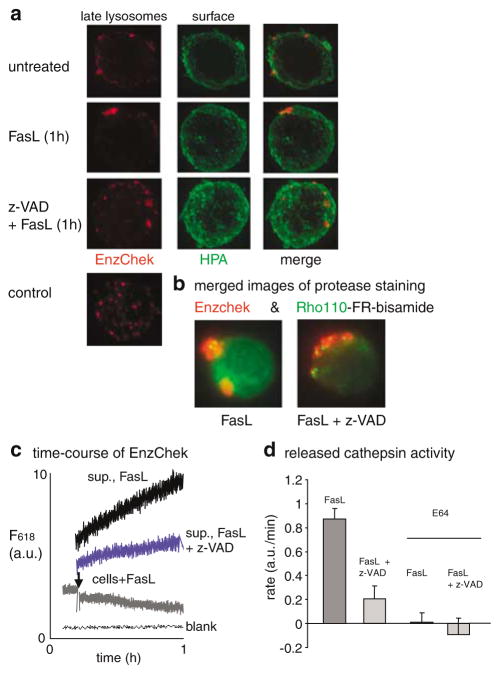
Subcellular organelles such as mitochondria, endoplasmic reticulum (ER) and the Golgi complex are involved in the progression of the cell death programme. We report here that soon after ligation of Fas (CD95/Apo1) in type II cells, elements of the Golgi complex intermix with mitochondria. This mixing follows centrifugal dispersal of secretory membranes and reflects a global alteration of membrane traffic. Activation of apical caspases is instrumental for promoting the dispersal of secretory organelles, since caspase inhibition blocks the outward movement of Golgi-related endomembranes and reduces their mixing with mitochondria. Caspase inhibition also blocks the FasL-induced secretion of intracellular proteases from lysosomal compartments, outlining a novel aspect of death receptor signalling via apical caspases. Thus, our work unveils that Fas ligand-mediated apoptosis induces scrambling of mitochondrial and secretory organelles via a global alteration of membrane traffic that is modulated by apical caspases.
Figures
References
-
- Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10:26–35. - PubMed
-
- Chipuk JE, Green DR. Do inducers of apoptosis trigger caspase-independent cell death? Nat Rev Mol Cel Biol. 2005;6:268–275. - PubMed
-
- Cristea IM, Degli Esposti M. Membrane lipids and cell death: an overview. Chem Phys Lipids. 2004;129:133–160. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
