

AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex
- PMID: 17015473
- PMCID: PMC1636878
- DOI: 10.1128/MCB.00597-06
AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex
Abstract
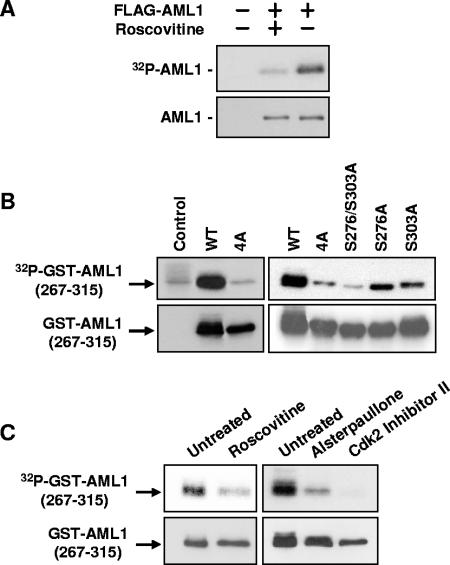
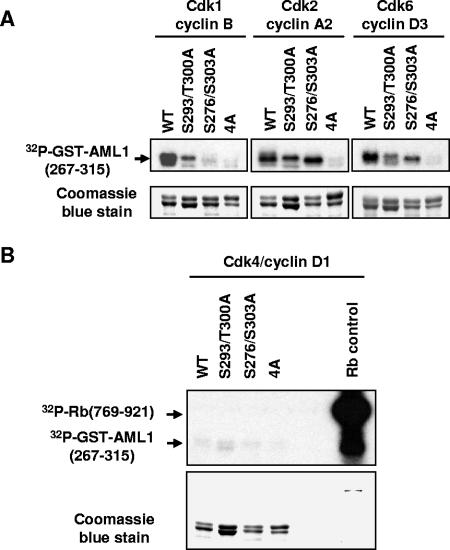
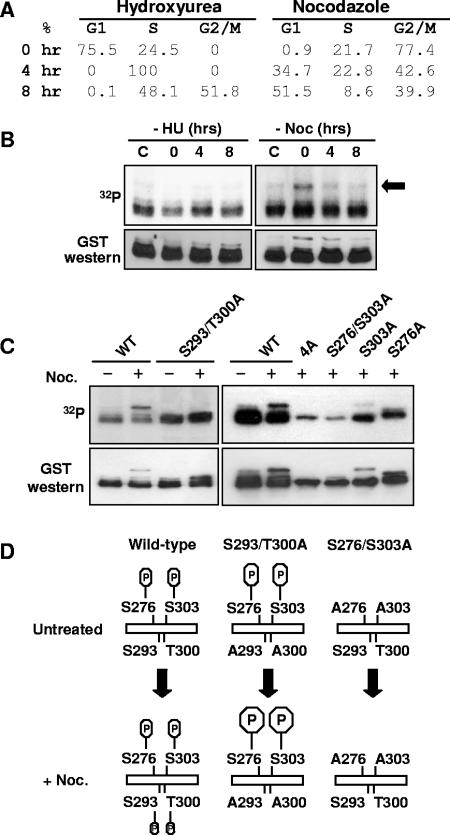
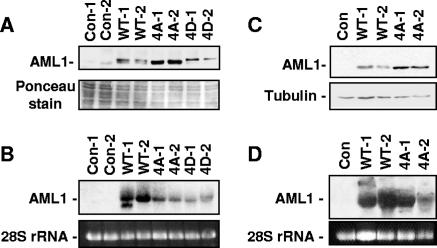
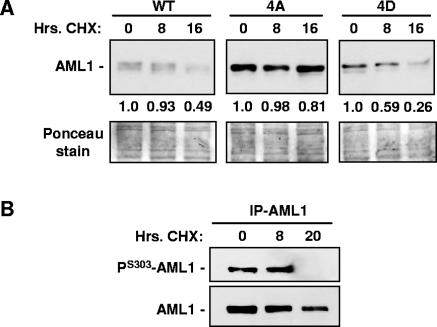
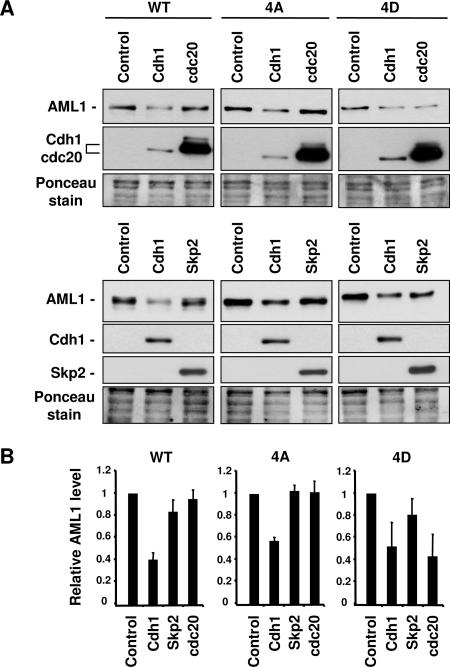
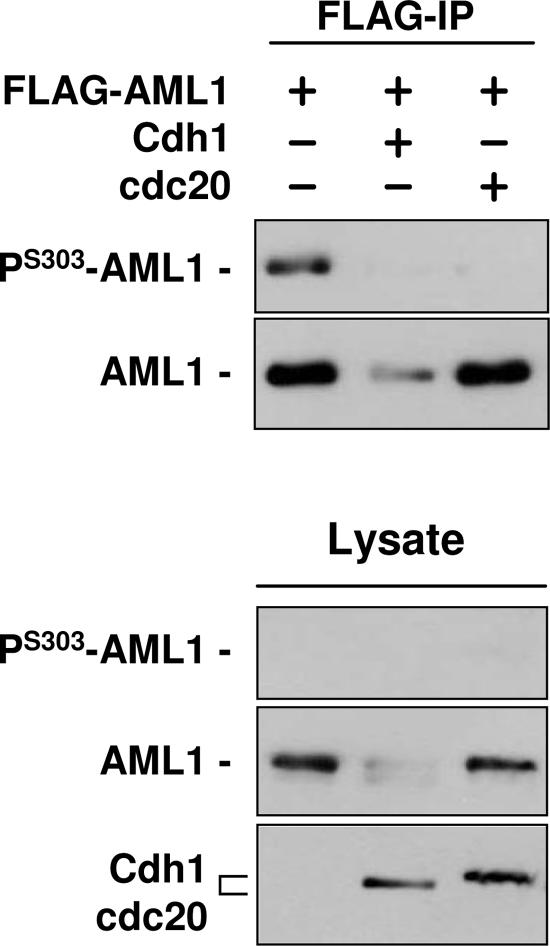
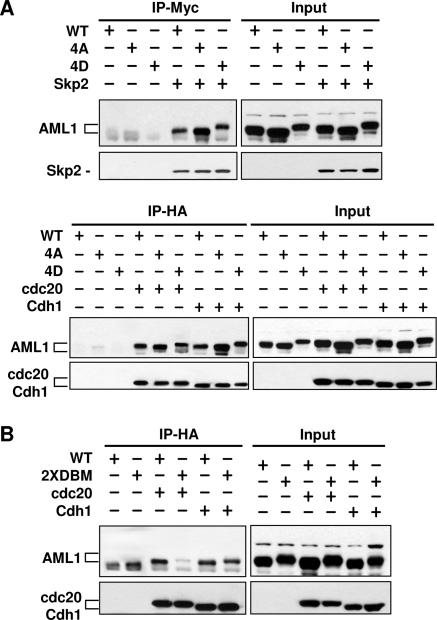
AML1 (RUNX1) regulates hematopoiesis, angiogenesis, muscle function, and neurogenesis. Previous studies have shown that phosphorylation of AML1, particularly at serines 276 and 303, affects its transcriptional activation. Here, we report that phosphorylation of AML1 serines 276 and 303 can be blocked in vivo by inhibitors of the cyclin-dependent kinases (CDKs) Cdk1 and Cdk2. Furthermore, these residues can be phosphorylated in vitro by purified Cdk1/cyclin B and Cdk2/cyclin A. Mutant AML1 protein which cannot be phosphorylated at these sites (AML1-4A) is more stable than wild-type AML1. AML-4A is resistant to degradation mediated by Cdc20, one of the substrate-targeting subunits of the anaphase-promoting complex (APC). However, Cdh1, another targeting subunit used by the APC, can mediate the degradation of AML1-4A. A phospho-mimic protein, AML1-4D, can be targeted by Cdc20 or Cdh1. These observations suggest that both Cdc20 and Cdh1 can target AML1 for degradation by the APC but that AML1 phosphorylation may affect degradation mediated by Cdc20-APC to a greater degree.
Figures


References
-
- Bernardin, F., and A. D. Friedman. 2002. AML1 stimulates G1 to S progression via its transactivation domain. Oncogene 21:3247-3252. - PubMed
-
- Bernardin-Fried, F., T. Kummalue, S. Leijen, M. I. Collector, K. Ravid, and A. D. Friedman. 2004. AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J. Biol. Chem. 279:15678-15687. - PubMed
-
- Biggs, J. R., Y. Zhang, L. F. Peterson, M. Garcia, D. E. Zhang, and A. S. Kraft. 2005. Phosphorylation of AML1/RUNX1 regulates its degradation and nuclear matrix association. Mol. Cancer Res. 3:391-401. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous