

The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported
- PMID: 17015493
- PMCID: PMC2910078
- DOI: 10.1542/peds.2006-0588
The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported
Erratum in
- Pediatrics. 2007 Oct;120(4):936
Abstract
Objective: Juvenile GM2 gangliosidosis is a group of inherited neurodegenerative diseases caused by deficiency of lysosomal beta-hexosaminidase resulting in GM2 ganglioside accumulation in brain. The purpose of this study was to delineate the natural history of the condition and identify genotype-phenotype correlations that might be helpful in predicting the course of the disease in individual patients.
Methods: A cohort of 21 patients with juvenile GM2 gangliosidosis, 15 with the Tay-Sachs variant and 6 with the Sandhoff variant, was studied prospectively in 2 centers. Our experience was compared with previously published reports on 134 patients. Information about clinical features, beta-hexosaminidase enzyme activity, and mutation analysis was collected.
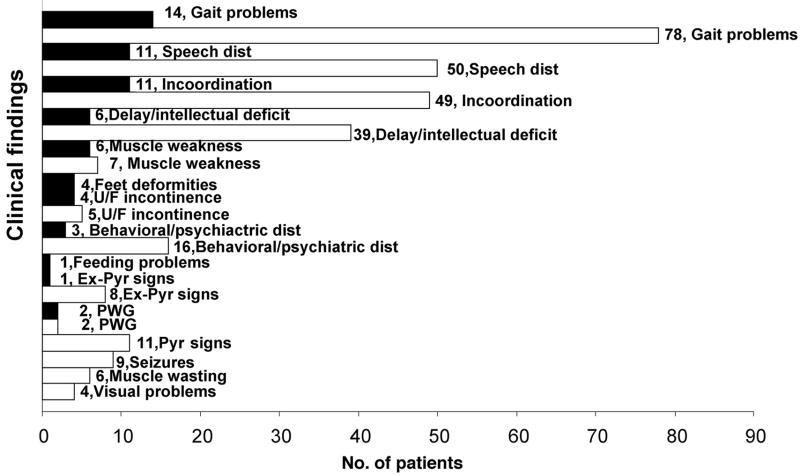
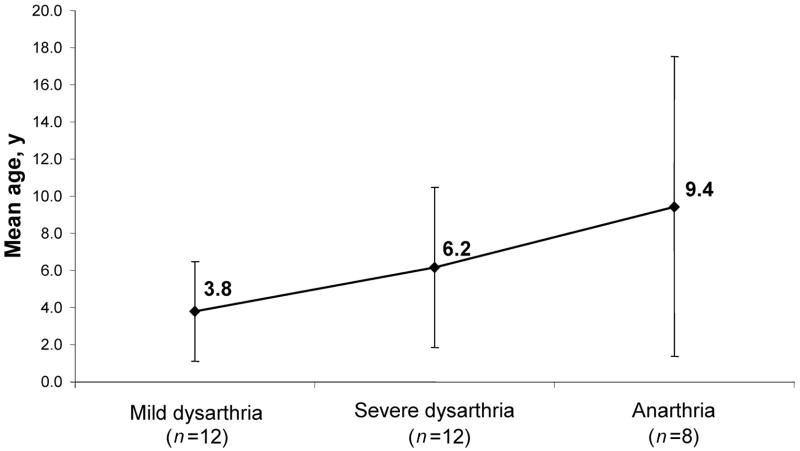
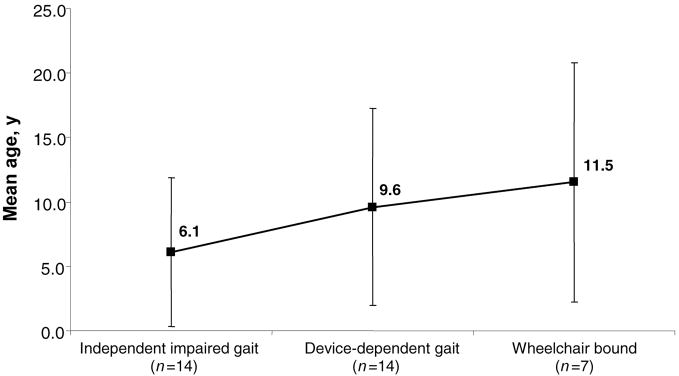
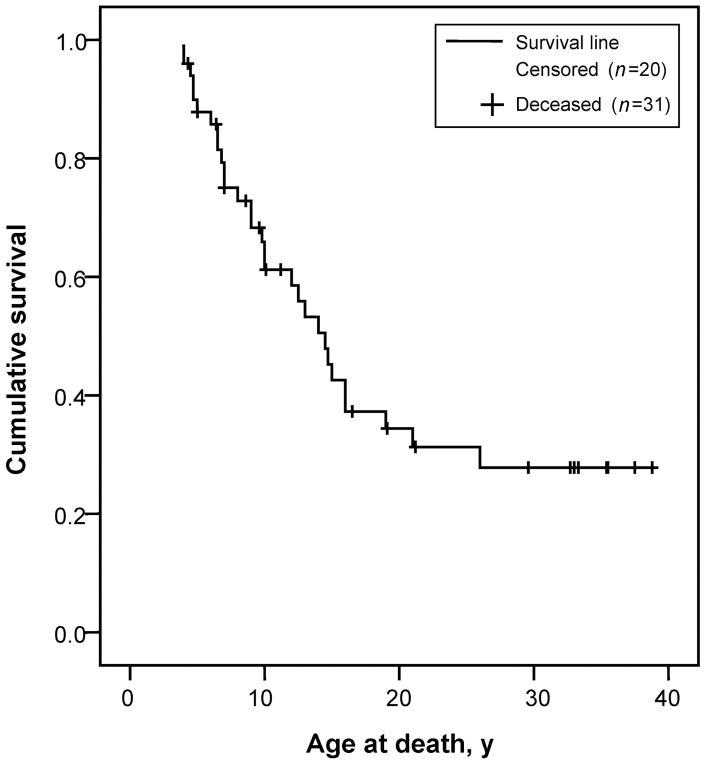
Results: In our cohort of patients, the mean (+/-SD) age of onset of symptoms was 5.3 +/- 4.1 years, with a mean follow-up time of 8.4 years. The most common symptoms at onset were gait disturbances (66.7%), incoordination (52.4%), speech problems (28.6%), and developmental delay (28.6%). The age of onset of gait disturbances was 7.1 +/- 5.6 years. The mean time for progression to becoming wheelchair-bound was 6.2 +/- 5.5 years. The mean age of onset of speech problems was 7.0 +/- 5.6 years, with a mean time of progression to anarthria of 5.6 +/- 5.3 years. Muscle wasting (10.6 +/- 7.4 years), proximal weakness (11.1 +/- 7.7 years), and incontinence of sphincters (14.6 +/- 9.7 years) appeared later in the course of the disease. Psychiatric disturbances and neuropathy were more prevalent in patients with the Sandhoff variant than in those with the Tay-Sachs variant. However, dysphagia, sphincter incontinence, and sleep problems occurred earlier in those with the Tay-Sachs variant. Cerebellar atrophy was the most common finding on brain MRI (52.9%). The median survival time among the studied and reviewed patients was 14.5 years. The genotype-phenotype correlation revealed that in patients with the Tay-Sachs variant, the presence of R178H and R499H mutations was predictive of an early onset and rapidly progressive course. The presence of either G269S or W474C mutations was associated with a later onset of symptoms along with a more slowly progressive disease course.
Conclusions: Juvenile GM2 gangliosidosis is clinically heterogeneous, not only in terms of age of onset and clinical features but also with regard to the course of the disease. In general, the earlier the onset of symptoms, the more rapidly the disease progresses. The Tay-Sachs and Sandhoff variants differed somewhat in the frequency of specific clinical characteristics. Speech deterioration progressed more rapidly than gait abnormalities in both the Tay-Sachs variant and Sandhoff variant groups. Among patients with the Tay-Sachs variant, the HEXA genotype showed a significant correlation with the clinical course.
Figures
References
-
- Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta. 1999;1455:105–138. - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. - PubMed
-
- Kaback MM. Population-based genetic screening for reproductive counseling: the Tay-Sachs disease model. Eur J Pediatr. 2000;159(suppl 3):S192–S195. - PubMed
-
- Kaback MM, Zeiger RS, Reynolds LW, Sonneborn M. Approaches to the control and prevention of Tay-Sachs disease. Prog Med Genet. 1974;10:103–134. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
