

Analysis of protein sequence and interaction data for candidate disease gene prediction
- PMID: 17020920
- PMCID: PMC1636487
- DOI: 10.1093/nar/gkl707
Analysis of protein sequence and interaction data for candidate disease gene prediction
Abstract
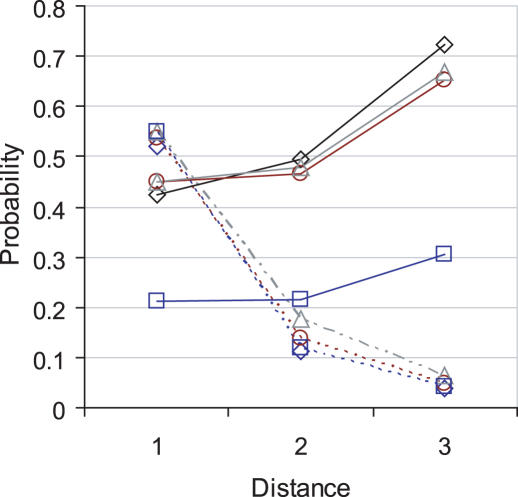
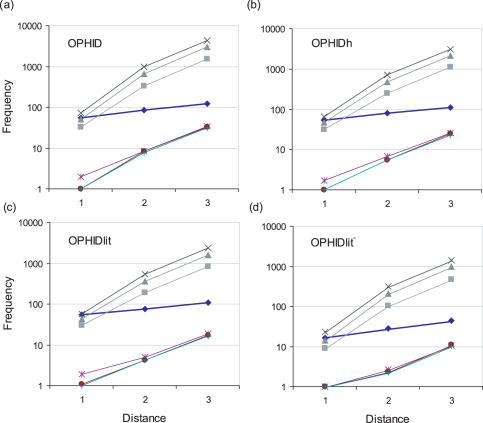
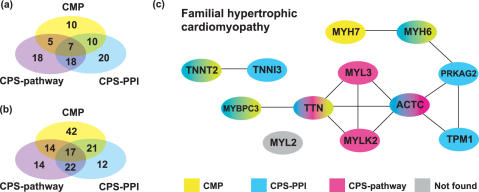
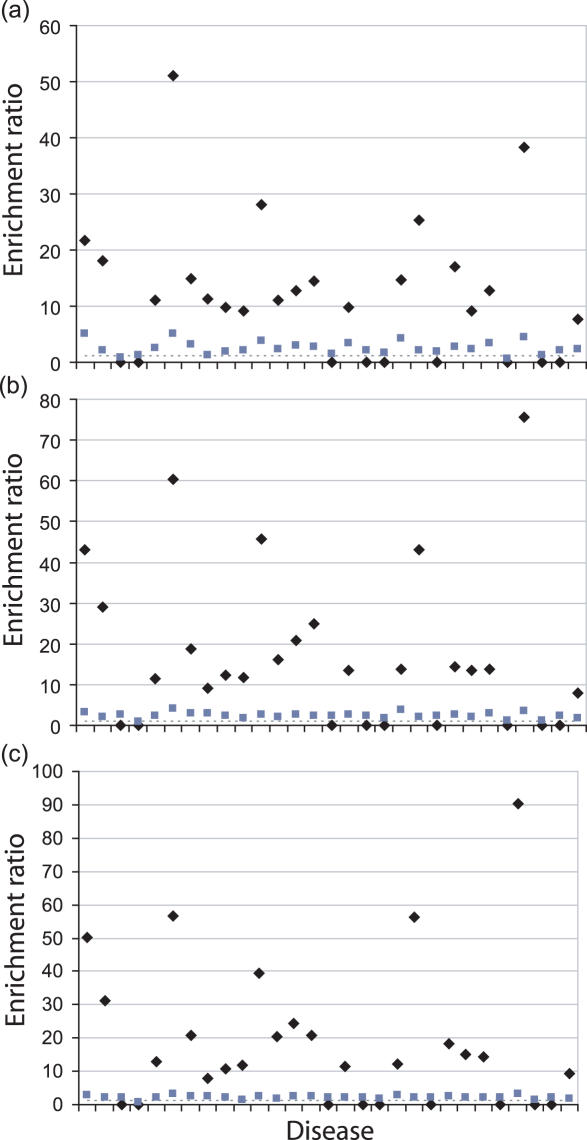
Linkage analysis is a successful procedure to associate diseases with specific genomic regions. These regions are often large, containing hundreds of genes, which make experimental methods employed to identify the disease gene arduous and expensive. We present two methods to prioritize candidates for further experimental study: Common Pathway Scanning (CPS) and Common Module Profiling (CMP). CPS is based on the assumption that common phenotypes are associated with dysfunction in proteins that participate in the same complex or pathway. CPS applies network data derived from protein-protein interaction (PPI) and pathway databases to identify relationships between genes. CMP identifies likely candidates using a domain-dependent sequence similarity approach, based on the hypothesis that disruption of genes of similar function will lead to the same phenotype. Both algorithms use two forms of input data: known disease genes or multiple disease loci. When using known disease genes as input, our combined methods have a sensitivity of 0.52 and a specificity of 0.97 and reduce the candidate list by 13-fold. Using multiple loci, our methods successfully identify disease genes for all benchmark diseases with a sensitivity of 0.84 and a specificity of 0.63. Our combined approach prioritizes good candidates and will accelerate the disease gene discovery process.
Figures
References
-
- Smyth D.J., Cooper J.D., Bailey R., Field S., Burren O., Smink L.J., Guja C., Ionescu-Tirgoviste C., Widmer B., Dunger D.B., et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nature Genet. 2006;38:617–619. - PubMed
-
- Perez-Iratxeta C., Bork P., Andrade M.A. Association of genes to genetically inherited diseases using data mining. Nature Genet. 2002;31:316–319. - PubMed
