

Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation
- PMID: 17021169
- PMCID: PMC6674618
- DOI: 10.1523/JNEUROSCI.1202-06.2006
Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation
Abstract
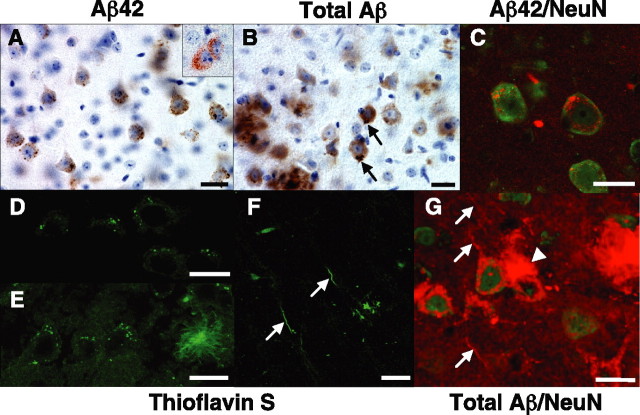
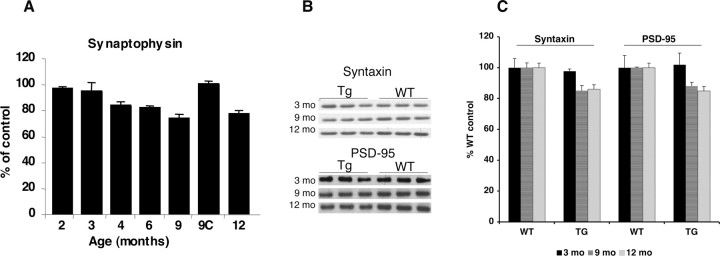
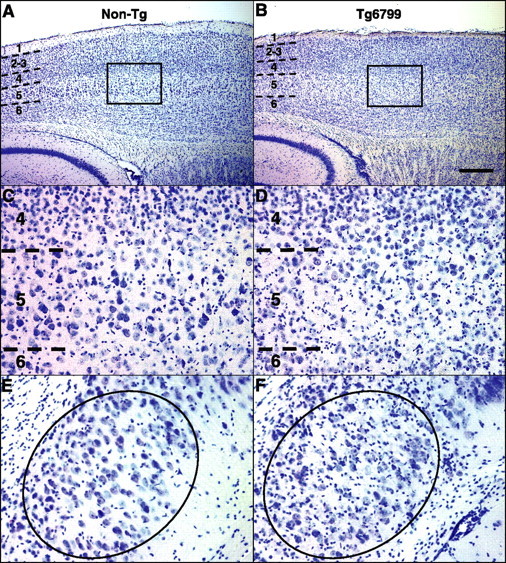
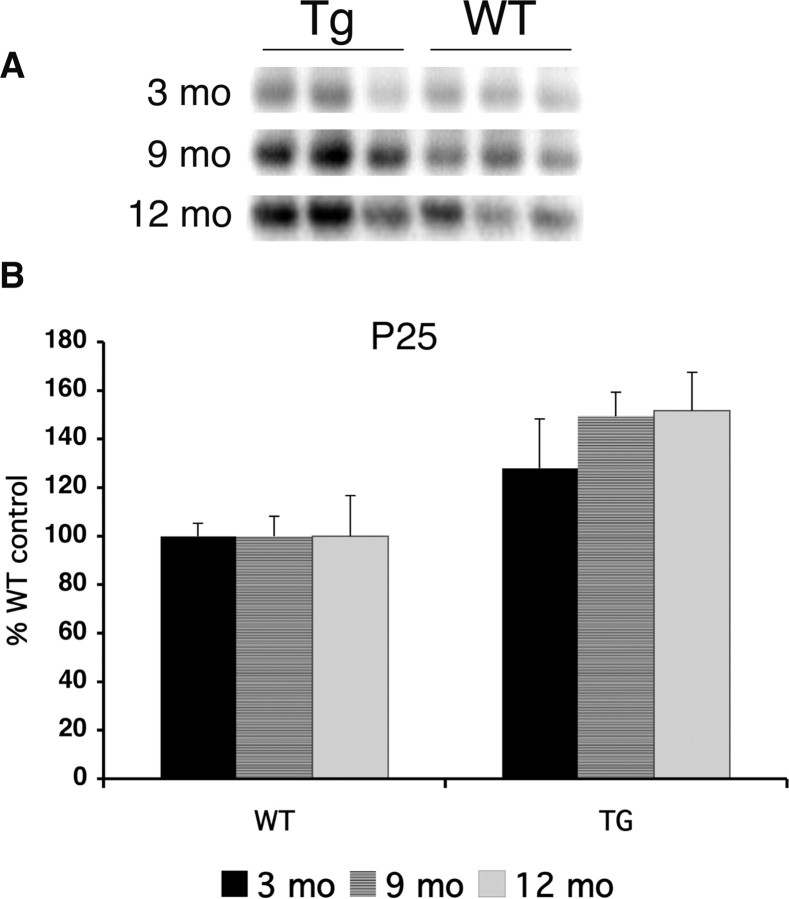
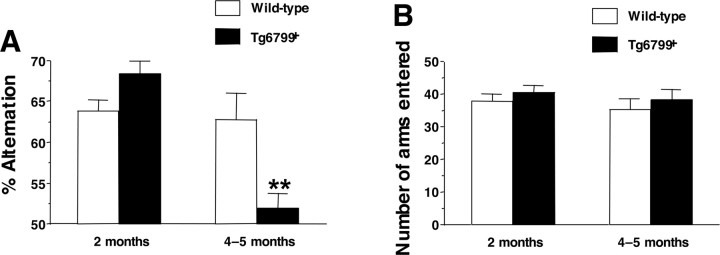
Mutations in the genes for amyloid precursor protein (APP) and presenilins (PS1, PS2) increase production of beta-amyloid 42 (Abeta42) and cause familial Alzheimer's disease (FAD). Transgenic mice that express FAD mutant APP and PS1 overproduce Abeta42 and exhibit amyloid plaque pathology similar to that found in AD, but most transgenic models develop plaques slowly. To accelerate plaque development and investigate the effects of very high cerebral Abeta42 levels, we generated APP/PS1 double transgenic mice that coexpress five FAD mutations (5XFAD mice) and additively increase Abeta42 production. 5XFAD mice generate Abeta42 almost exclusively and rapidly accumulate massive cerebral Abeta42 levels. Amyloid deposition (and gliosis) begins at 2 months and reaches a very large burden, especially in subiculum and deep cortical layers. Intraneuronal Abeta42 accumulates in 5XFAD brain starting at 1.5 months of age (before plaques form), is aggregated (as determined by thioflavin S staining), and occurs within neuron soma and neurites. Some amyloid deposits originate within morphologically abnormal neuron soma that contain intraneuronal Abeta. Synaptic markers synaptophysin, syntaxin, and postsynaptic density-95 decrease with age in 5XFAD brain, and large pyramidal neurons in cortical layer 5 and subiculum are lost. In addition, levels of the activation subunit of cyclin-dependent kinase 5, p25, are elevated significantly at 9 months in 5XFAD brain, although an upward trend is observed by 3 months of age, before significant neurodegeneration or neuron loss. Finally, 5XFAD mice have impaired memory in the Y-maze. Thus, 5XFAD mice rapidly recapitulate major features of AD amyloid pathology and may be useful models of intraneuronal Abeta42-induced neurodegeneration and amyloid plaque formation.
Figures
References
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. - PMC - PubMed
-
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem. 2001;8:301–308. - PubMed
-
- Bales KR, Dodart JC, DeMattos RB, Holtzman DM, Paul SM. Apolipoprotein E, amyloid, and Alzheimer disease. Mol Interv. 2002;2:363–375. - PubMed
-
- Biernat J, Mandelkow E-M, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren M, Goedert M, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. - PMC - PubMed
-
- Billings LM, Oddo S, Green KN, McGaugh JL, Laferla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases