

Biological systems modeling and analysis: a biomolecular technique of the twenty-first century
- PMID: 17028166
- PMCID: PMC2291792
Biological systems modeling and analysis: a biomolecular technique of the twenty-first century
Abstract
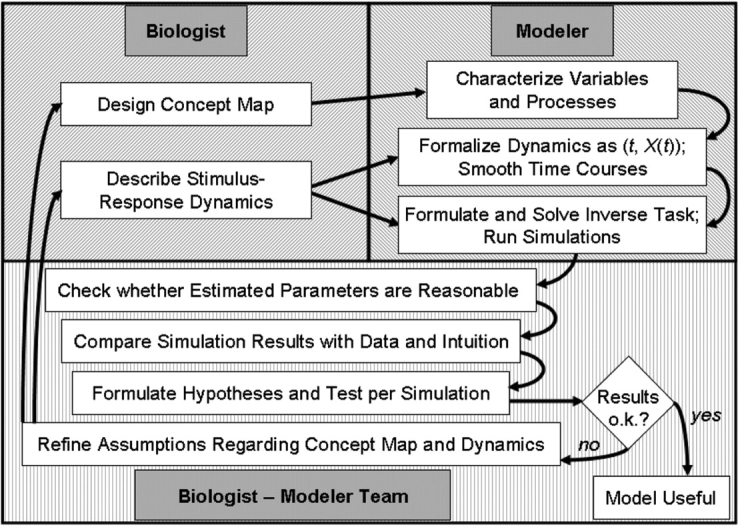
It is proposed that computational systems biology should be considered a biomolecular technique of the twenty-first century, because it complements experimental biology and bioinformatics in unique ways that will eventually lead to insights and a depth of understanding not achievable without systems approaches. This article begins with a summary of traditional and novel modeling techniques. In the second part, it proposes concept map modeling as a useful link between experimental biology and biological systems modeling and analysis. Concept map modeling requires the collaboration between biologist and modeler. The biologist designs a regulated connectivity diagram of processes comprising a biological system and also provides semi-quantitative information on stimuli and measured or expected responses of the system. The modeler converts this information through methods of forward and inverse modeling into a mathematical construct that can be used for simulations and to generate and test new hypotheses. The biologist and the modeler collaboratively interpret the results and devise improved concept maps. The third part of the article describes software, BST-Box, supporting the various modeling activities.
Figures














References
-
- Voit EO. Models-of-data and models-of processes in the post-genomic era. Special Issue in honor of John A. Jacquez. Math Biosci 2002;180:263–274. - PubMed
-
- Leicester HM. Development of Biochemical Concepts from Ancient to Modern Times. 1974, Cambridge, MA: Harvard University Press.
-
- Voit EO, Schwacke JH. Understanding through Modeling, in Konopka AK, Editor. Systems Biolog y: Principles, Methods, and Concepts, in press, 2006.
-
- Crampin EJ, Halstead M, Hunter P, Nielsen P, Noble D, Smith N, et al. Computational physiology and the Physiome Project. Exp Physiol 2004;89:1–26. - PubMed
-
- Westerhoff HV, Palsson BO. The evolution of molecular biology into systems biology. Nat Biotechnol 2004;22:1249–52. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources