

BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites
- PMID: 17030608
- PMCID: PMC1698544
- DOI: 10.1128/MCB.01099-06
BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites
Abstract
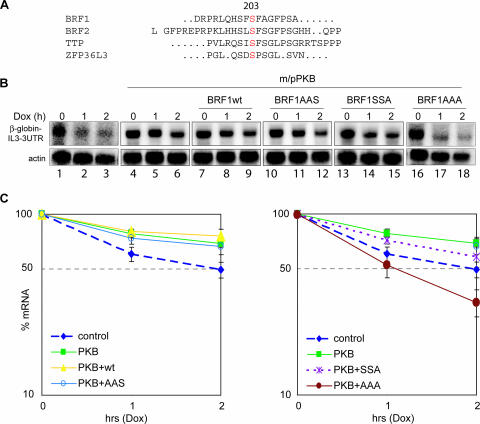
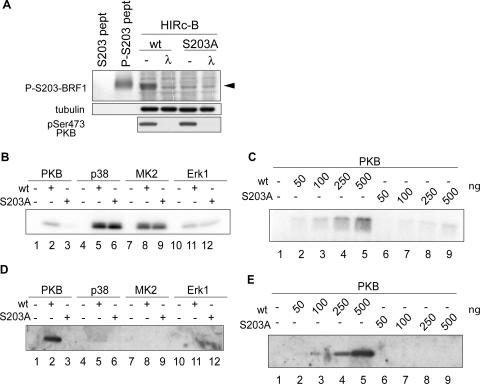
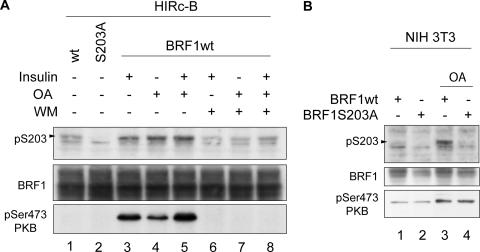
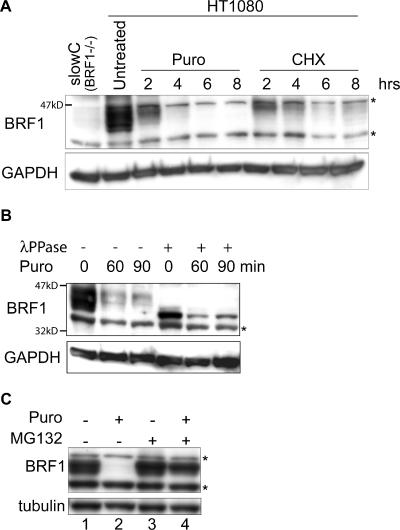
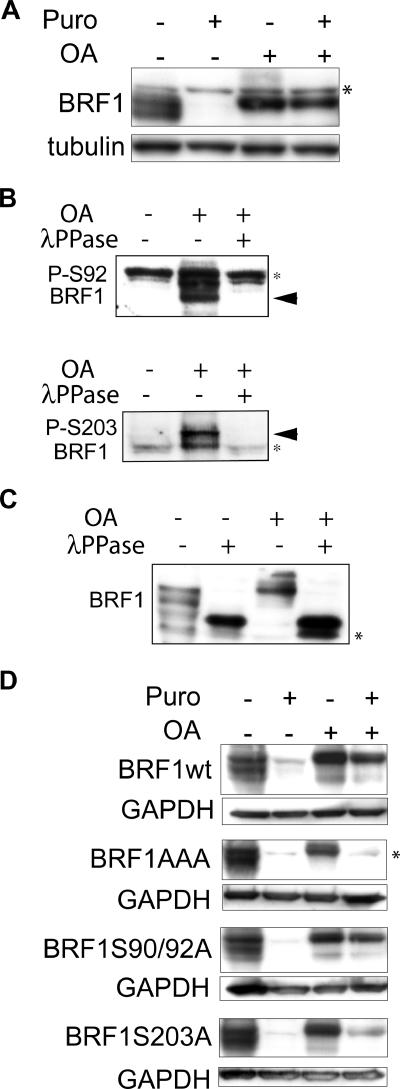
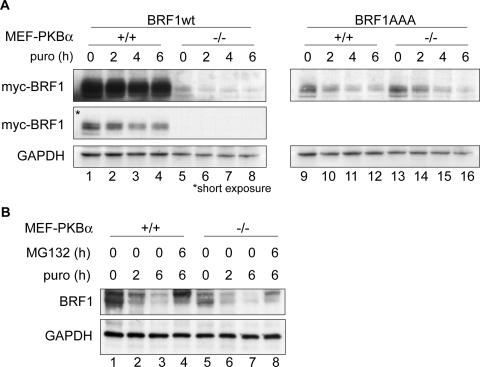
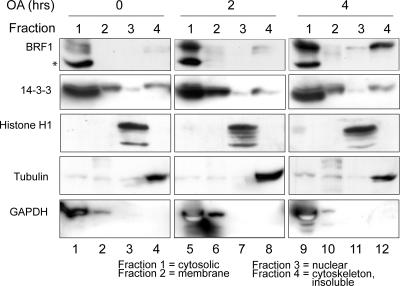
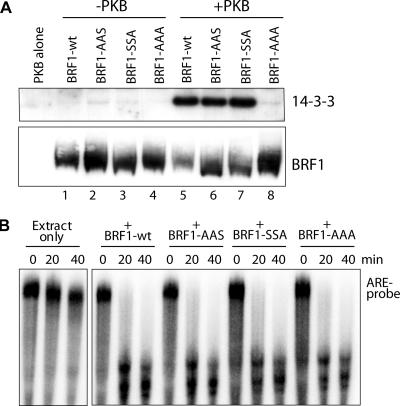
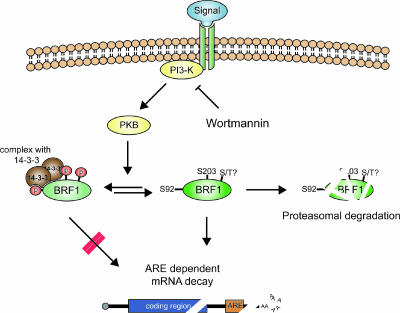
BRF1 posttranscriptionally regulates mRNA levels by targeting ARE-bearing transcripts to the decay machinery. We previously showed that protein kinase B (PKB) phosphorylates BRF1 at Ser92, resulting in binding to 14-3-3 and impairment of mRNA decay activity. Here we identify an additional regulatory site at Ser203 that cooperates in vivo with Ser92. In vitro kinase labeling and wortmannin sensitivity indicate that Ser203 phosphorylation is also performed by PKB. Mutation of both serines to alanine uncouples BRF1 from PKB regulation, leading to constitutive mRNA decay even in the presence of stabilizing signals. BRF1 protein is labile because of proteasomal degradation (half-life, <3 h) but becomes stabilized upon phosphorylation and is less stable in PKBalpha(-/-) cells. Surprisingly, phosphorylation-dependent protein stability is also regulated by Ser92 and Ser203, with parallel phosphorylation required at these sites. Phosphorylation-dependent binding to 14-3-3 is abolished only when both sites are mutated. Cell compartment fractionation experiments support a model in which binding to 14-3-3 sequesters BRF1 through relocalization and prevents it from executing its mRNA decay activity, as well as from proteasomal degradation, thereby maintaining high BRF1 protein levels that are required to reinstate decay upon dissipation of the stabilizing signal.
Figures
References
-
- Aitken, A., H. Baxter, T. Dubois, S. Clokie, S. Mackie, K. Mitchell, A. Peden, and E. Zemlickova. 2002. Specificity of 14-3-3 isoform dimer interactions and phosphorylation. Biochem. Soc. Trans. 30:351-360. - PubMed
-
- Andjelković, M., D. R. Alessi, R. Meier, A. Fernandez, N. J. Lamb, M. Frech, P. Cron, P. Cohen, J. M. Lucocq, and B. A. Hemmings. 1997. Role of translocation in the activation and function of protein kinase B. J. Biol. Chem. 272:31515-31524. - PubMed
-
- Baggs, J. E., and C. B. Green. 2003. Nocturnin, a deadenylase in Xenopus laevis retina: a mechanism for posttranscriptional control of circadian-related mRNA. Curr. Biol. 13:189-198. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous