

Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome
- PMID: 17033964
- PMCID: PMC1698567
- DOI: 10.1086/508474
Mutations of the mitochondrial holocytochrome c-type synthase in X-linked dominant microphthalmia with linear skin defects syndrome
Abstract
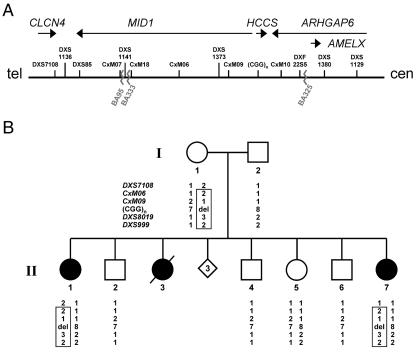
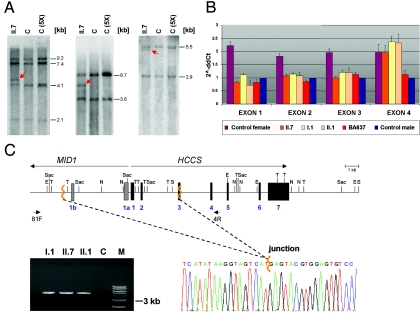
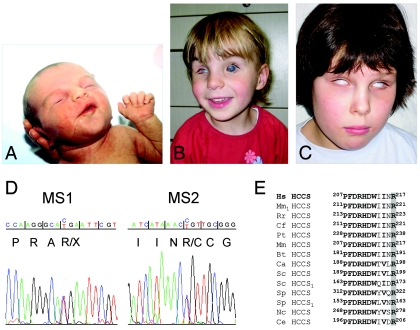
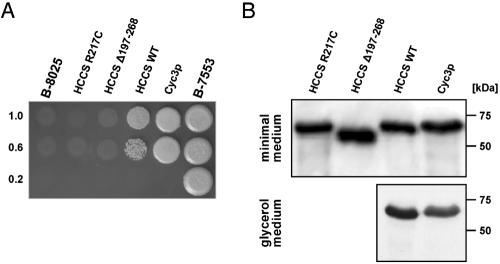
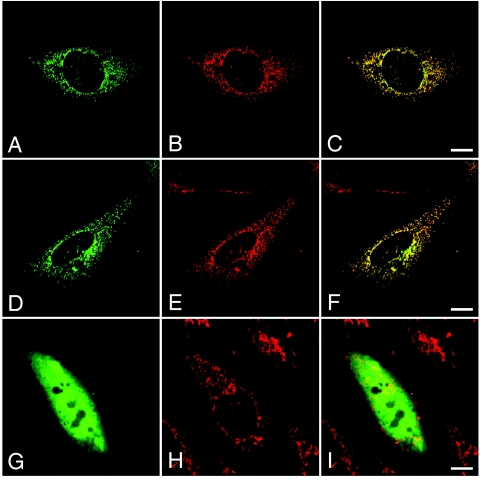
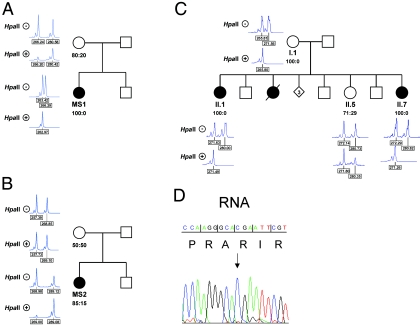
The microphthalmia with linear skin defects syndrome (MLS, or MIDAS) is an X-linked dominant male-lethal disorder almost invariably associated with segmental monosomy of the Xp22 region. In two female patients, from two families, with MLS and a normal karyotype, we identified heterozygous de novo point mutations--a missense mutation (p.R217C) and a nonsense mutation (p.R197X)--in the HCCS gene. HCCS encodes the mitochondrial holocytochrome c-type synthase that functions as heme lyase by covalently adding the prosthetic heme group to both apocytochrome c and c(1). We investigated a third family, displaying phenotypic variability, in which the mother and two of her daughters carry an 8.6-kb submicroscopic deletion encompassing part of the HCCS gene. Functional analysis demonstrates that both mutant proteins (R217C and Delta 197-268) were unable to complement a Saccharomyces cerevisiae mutant deficient for the HCCS orthologue Cyc3p, in contrast to wild-type HCCS. Moreover, ectopically expressed HCCS wild-type and the R217C mutant protein are targeted to mitochondria in CHO-K1 cells, whereas the C-terminal-truncated Delta 197-268 mutant failed to be sorted to mitochondria. Cytochrome c, the final product of holocytochrome c-type synthase activity, is implicated in both oxidative phosphorylation (OXPHOS) and apoptosis. We hypothesize that the inability of HCCS-deficient cells to undergo cytochrome c-mediated apoptosis may push cell death toward necrosis that gives rise to severe deterioration of the affected tissues. In summary, we suggest that disturbance of both OXPHOS and the balance between apoptosis and necrosis, as well as the X-inactivation pattern, may contribute to the variable phenotype observed in patients with MLS.
Figures
References
Web Resources
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for HCCS [accession number NM_005333], RPP30 [accession number NM_006413], and HCCS cDNA clone DKFZp779I1858 [accession number CR749578])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MLS phenotypes)
-
- RZPD German Resource Center for Genome Research, http://www.rzpd.de/
References
-
- Morleo M, Pramparo T, Perone L, Gregato G, Le Caignec C, Mueller RF, Ogata T, Raas-Rothschild A, de Blois MC, Wilson LC, Zaidman G, Zuffardi O, Ballabio A, Franco B (2005) Microphthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic, and molecular characterization of 11 cases. Am J Med Genet A 137:190–198 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
