

Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats
- PMID: 17034580
- PMCID: PMC1942064
- DOI: 10.1111/j.1365-2249.2006.03198.x
Complement membrane attack is required for endplate damage and clinical disease in passive experimental myasthenia gravis in Lewis rats
Abstract
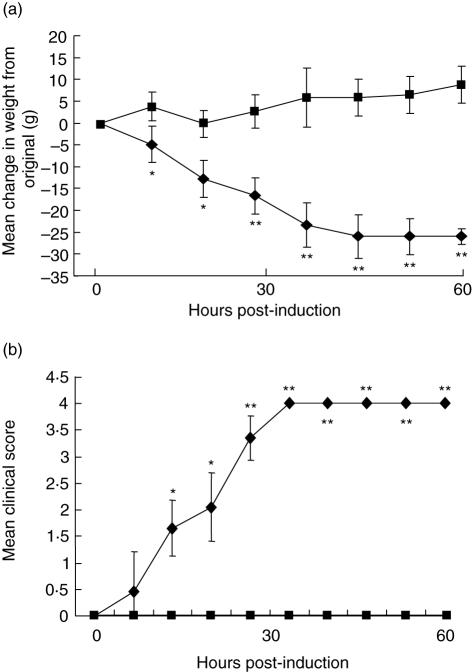
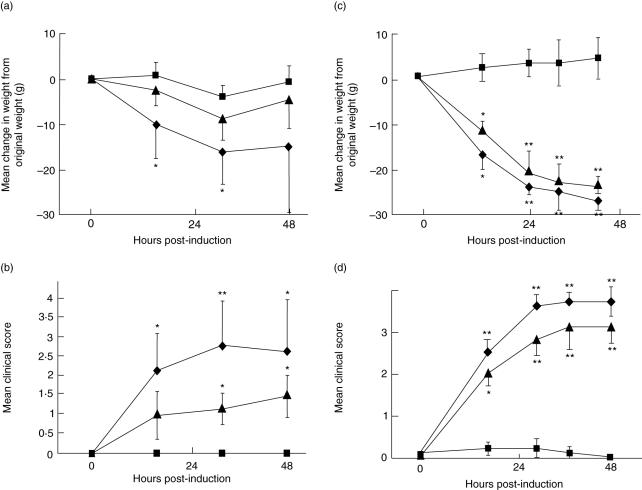
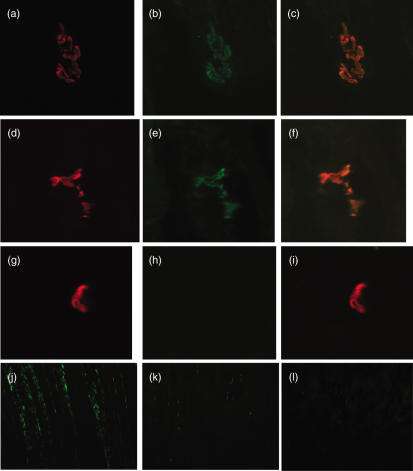
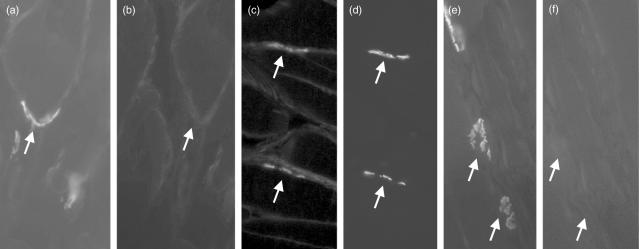
Myasthenia gravis (MG) is a debilitating and potentially fatal neuromuscular disease characterized by the generation of autoantibodies reactive with nicotinic acetylcholine receptors (AChR) that cause loss of AChR from the neuromuscular endplate with resultant failure of neuromuscular transmission. A role for complement (C) in the pathology of human MG has been suggested based upon identification of C activation products in plasma and deposited at the endplate in MG. In the rat model, experimental autoimmune MG (EAMG), C depletion or inhibition restricts clinical disease, further implicating C in pathology. The mechanisms by which C activation drives pathology in MG and EAMG are unclear. Here we provide further evidence implicating C and specifically the membrane attack complex (MAC) in the Lewis rat passive EAMG model of MG. Rats deficient in C6, an essential component of the MAC, were resistant to disease induction and endplate destruction was reduced markedly compared to C6-sufficient controls. After reconstitution with C6, disease severity and endplate destruction in the C6-deficient rats was equivalent to that in controls. The data confirm the essential role of the MAC in the destruction of the endplate in EAMG and raise the prospect of specific MAC inhibition as an alternative therapy in MG patients resistant to conventional treatments.
Figures

References
-
- Hughes BW, De Casillas MLM, Kaminski HJ. Pathophysiology of myasthenia gravis. Semin Neurol. 2004;24:21–30. - PubMed
-
- Lindstrom J. Acetylcholine receptors and myasthenia. Muscle Nerve. 2000;23:453–77. - PubMed
-
- Lindstrom J. Nicotinic acetylcholine receptors of muscles and nerves. Comparison of their structures, functional roles and vulnerability to pathology. Ann NY Acad Sci. 2003;998:41–52. - PubMed
-
- Newsom-Davis J, Pinching AJ, Vincent A, Wilson SG. Function of circulating antibody to acetylcholine receptor in myasthenia gravis: investigation by plasma exchange. Neurology. 1978;28:266–72. - PubMed
-
- Nastuk WL, Plescia OJ, Osserman KE. Changes in serum complement activity in patients with myasthenia gravis. Proc Soc Exp Biol Med. 1960;105:177–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
