

Brugada syndrome
- PMID: 17038146
- PMCID: PMC1978482
- DOI: 10.1111/j.1540-8159.2006.00507.x
Brugada syndrome
Abstract
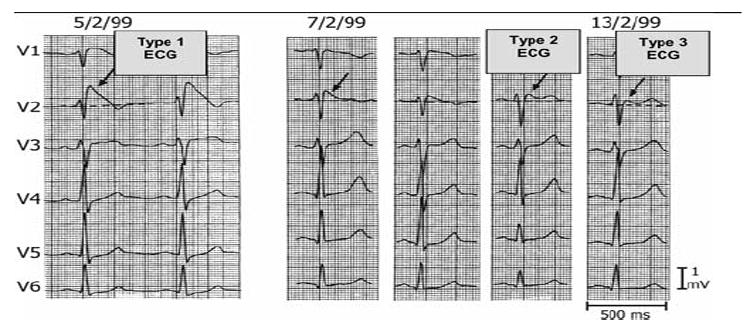
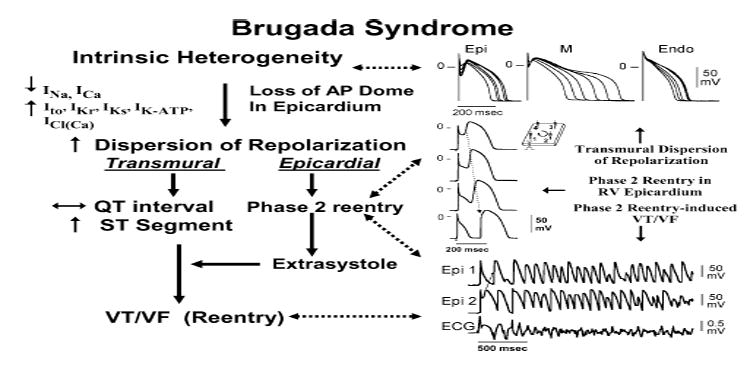
First introduced as a new clinical entity in 1992, the Brugada syndrome is associated with a relatively high risk of sudden death in young adults, and occasionally in children and infants. Recent years have witnessed a striking proliferation of papers dealing with the clinical and basic aspects of the disease. Characterized by a coved-type ST-segment elevation in the right precordial leads of the electrocardiogram (ECG), the Brugada syndrome has a genetic basis that thus far has been linked only to mutations in SCN5A, the gene that encodes the alpha-subunit of the sodium channel. The Brugada ECG is often concealed, but can be unmasked or modulated by a number of drugs and pathophysiological states including sodium channel blockers, a febrile state, vagotonic agents, tricyclic antidepressants, as well as cocaine and propranolol intoxication. Average age at the time of initial diagnosis or sudden death is 40 +/- 22, with the youngest patient diagnosed at 2 days of age and the oldest at 84 years. This review provides an overview of the clinical, genetic, molecular, and cellular aspects of the Brugada syndrome, incorporating the results of two recent consensus conferences. Controversies with regard to risk stratification and newly proposed pharmacologic strategies are discussed.
Figures















References
-
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. - PubMed
-
- Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada syndrome. A decade of progress. Circ Res. 2002;91:1114–1119. - PubMed
-
- Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Eur Heart J. 2002;23:1648–1654. - PubMed
-
- Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation. 2002;106:2514–2519. - PubMed
-
- Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome. Report of the second consensus conference. Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
