

Review
doi: 10.1038/nrmicro1532.
Epub 2006 Oct 9.
Genomics meets HIV-1
Affiliations
- PMID: 17041633
- PMCID: PMC2043131
- DOI: 10.1038/nrmicro1532
Item in Clipboard
Review
Genomics meets HIV-1
Nat Rev Microbiol.
2006 Nov.
Abstract
Genomics is now a core element in the effort to develop a vaccine against HIV-1. Thanks to unprecedented progress in high-throughput genotyping and sequencing, in knowledge about genetic variation in humans, and in evolutionary genomics, it is finally possible to systematically search the genome for common genetic variants that influence the human response to HIV-1. The identification of such variants would help to determine which aspects of the response to the virus are the most promising targets for intervention. However, a key obstacle to progress remains the scarcity of appropriate human cohorts available for genomic research.
Conflict of interest statement
Competing interests statement
The authors declare no competing financial interests.
Figures
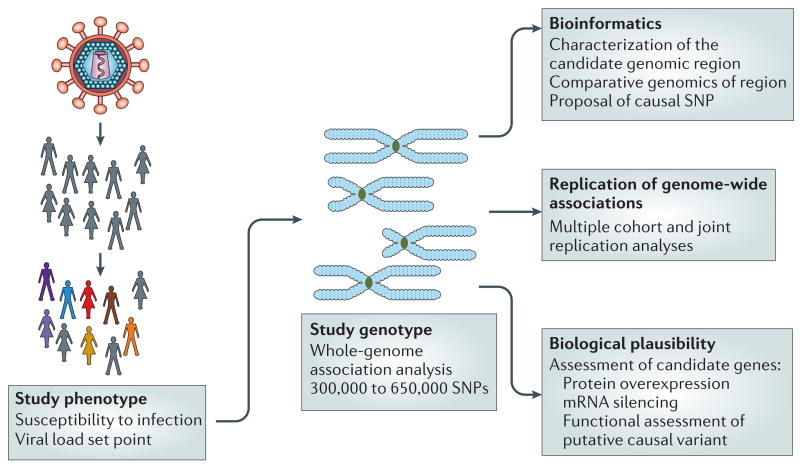
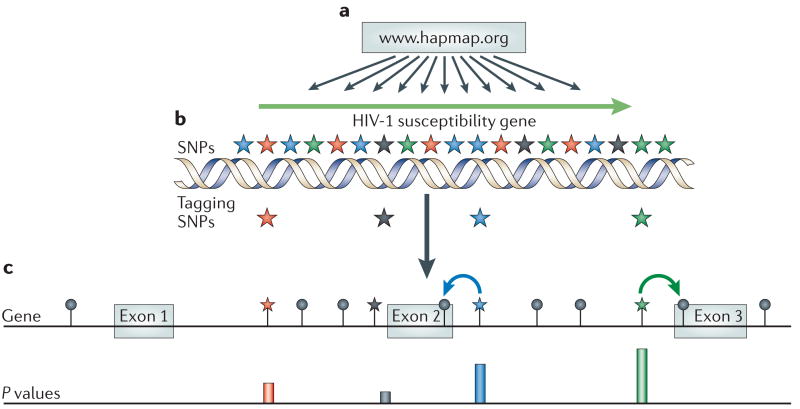
The figure shows the steps needed to define a study phenotype and genotype, and the downstream procedures used to prioritize candidate genes to be investigated by genetic and functional approaches. The study phenotype(s) should be robust, that is, consistent, and should represent a characteristic of the individual that can be measured precisely. Examples include susceptibility to infection or viral load set point. Genotyping should take into consideration the specific population studied, as the number of polymorphic markers (single-nucleotide polymorphisms; SNPs) that require analysis differs for African or non-African populations. After the identification of genetic markers that are associated with the phenotype, a comprehensive bioinformatics and genetic analysis of the genomic region is done to identify causal candidate variants associated with the marker. Also, replication studies in other human cohorts, analysis in the context of other HIV-1 infection endpoints or phenotypes, and dedicated biological studies dictated by the nature of the genes that are mapped and their plausible mechanism of action are used to prioritize appropriate candidate genes.
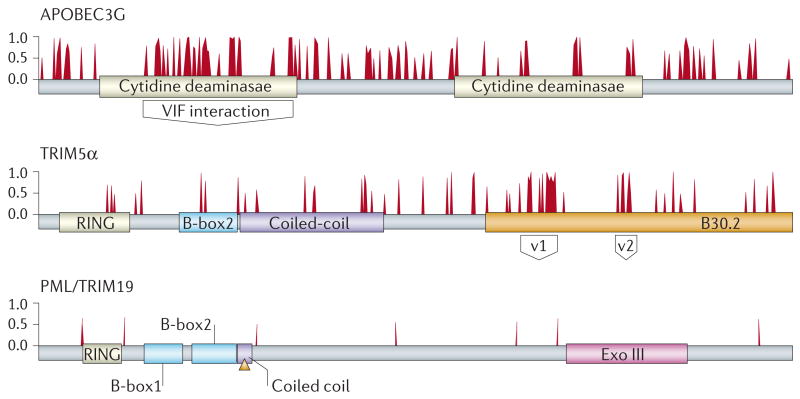
This analysis used a comprehensive set of sequences that are representative of the primate lineages — new world monkeys, old world monkeys, gibbons and apes — to identify signs of positive and of negative selection in protein domains. The three proteins depicted represent different patterns of evolution in primates. The antiretroviral protein APOBEC3G is characterized by many residues that are under positive selective pressure, with a large cluster that delineates the virion infectivity factor (Vif)-interaction domain. These amino acids contribute to discrimination among the various Vif proteins of HIV and different SIVs. A second antiretroviral protein, TRIM5α (tripartite motif 5α) has a patch of positively selected residues that defines the variable 1 region (v1) and variable 2 region (v2) and that carry the capsid recognition specificity. The variable regions of TRIM5α might have evolved independently to recognize various retroviruses. By contrast, there are no residues under positive selective pressure in the PML/TRIM19 protein, which is also proposed to have antiviral properties. Therefore, the effect of PML/TRIM19 might be indirect. Failure to identify a signature of positive selection militates against a more direct role for this protein in antiviral defence, because it would be expected that prolonged contact with several pathogens over long evolutionary time periods would have resulted in signatures of positive selection indicative of a genetic conflict. Bars represent amino acids that are predicted to be under positive selection in a grading of posterior probability from 0 to 1. The various protein domains are indicated. Adapted with permission from Ref. .
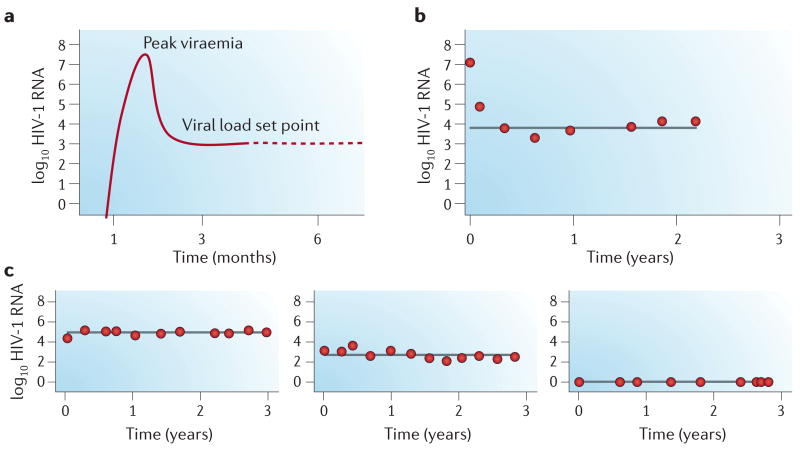
a | Soon after infection, HIV replicates to high levels (peak viraemia). Host factors then exert their effects to limit viral replication to a post-acute set-point level of plasma viraemia. b | The dynamics of establishment of the viral load set point in an individual presenting with HIV primary infection are shown. c | Viraemia data from three individuals are shown, characterized by post-acute viral load set points at 5, 3 and <2 log10 HIV-1 RNA copies per millilitre — fully stable over 3 years. Without these reliable patterns of inter-individual variation, genetics would have little to contribute to the analysis of viral load set point.
References
-
- Ioannidis JP. Commentary: grading the credibility of molecular evidence for complex diseases. Int J Epidemiol. 2006;35:572–578. - PubMed
-
- O'Brien SJ, Nelson GW. Human genes that limit AIDS. Nature Genet. 2004;36:565–574. - PubMed
-
- Telenti A, Bleiber G. Host genetics of HIV-1 susceptibility. Future Virol. 2006;1:55–70.
-
- Todd JA. Statistical false positive or true disease pathway? Nature Genet. 2006;38:731–733. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
