

Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2
- PMID: 17041889
- PMCID: PMC2799897
- DOI: 10.1002/humu.9461
Recessive arrhythmogenic right ventricular dysplasia due to novel cryptic splice mutation in PKP2
Abstract
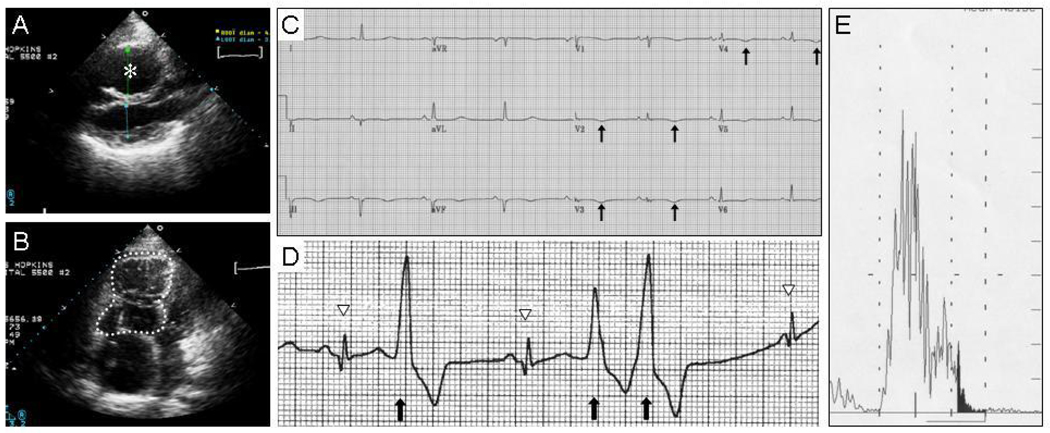
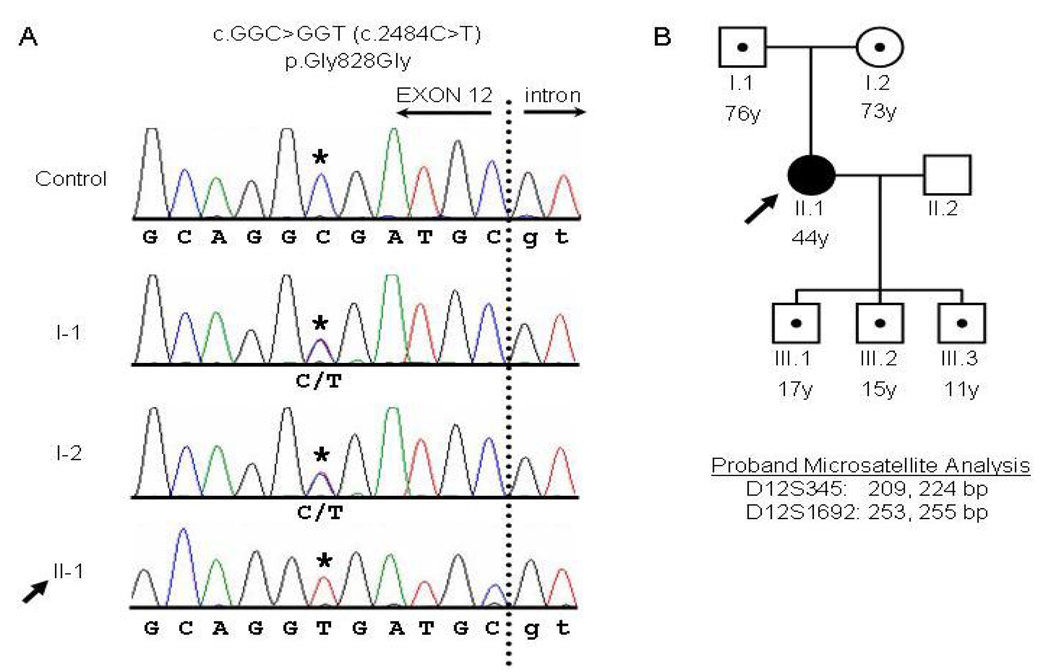
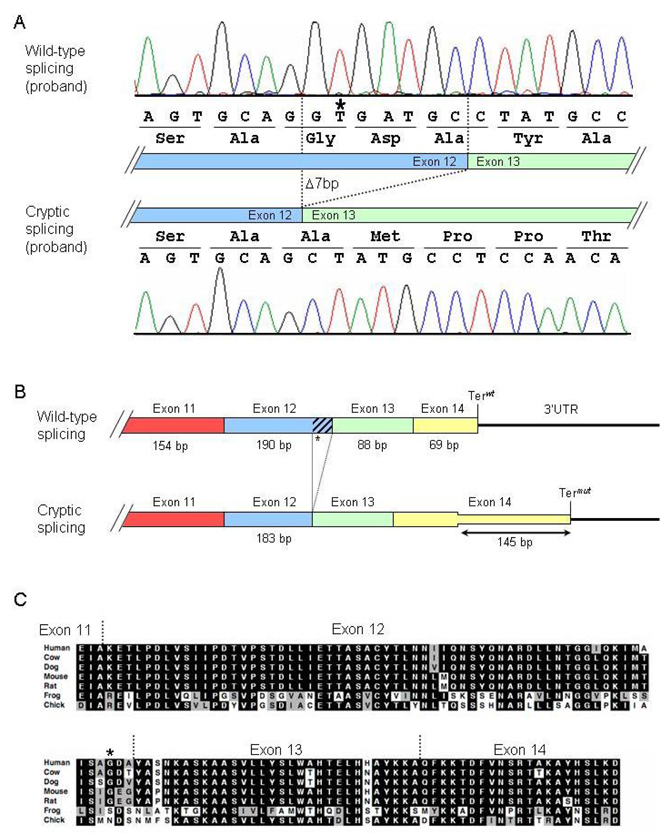
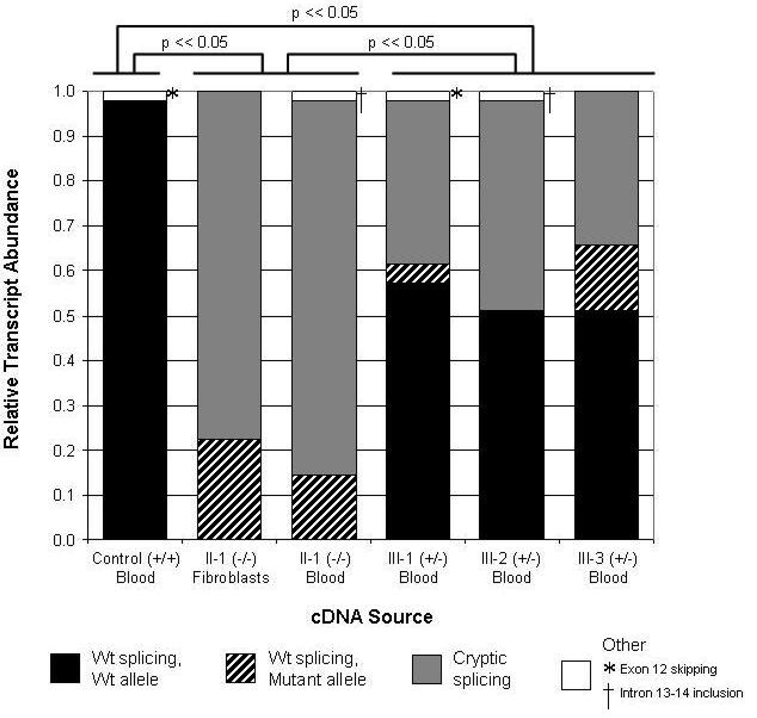
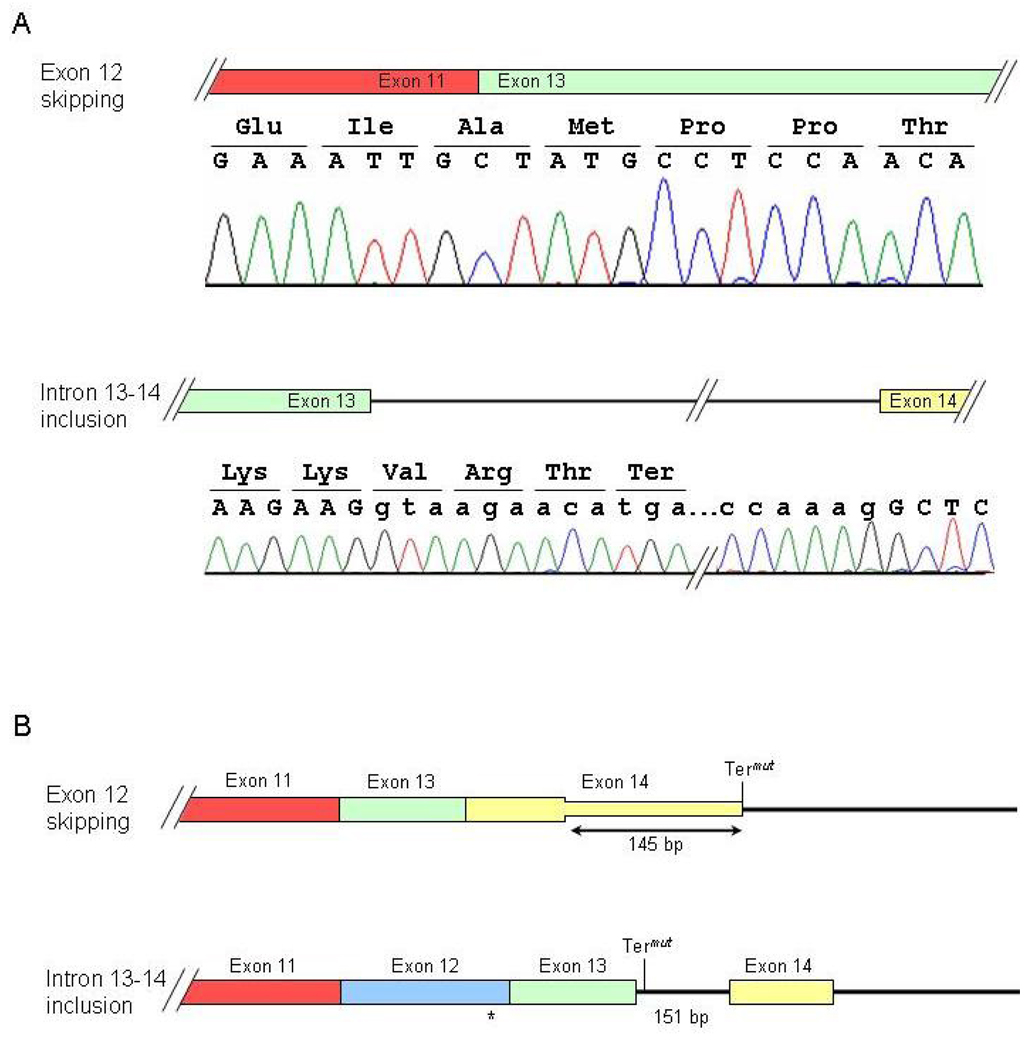
Arrhythmogenic right ventricular dysplasia (ARVD) is a genetic disorder resulting in fibro-fatty replacement of right ventricular myocytes and consequent ventricular arrhythmias. Heterozygous mutations in PKP2 encoding plakophilin-2 have previously been reported to cause dominant ARVD with reduced penetrance. We report the first case of recessive ARVD caused by mutations in PKP2. Candidate gene analysis in a typical proband with this disorder identified a novel homozygous mutation in PKP2 (c.[2484C>T]+[2484C>T]), which is predicted to be translationally silent (p.Gly828). Analysis of the proband's mRNA, however, shows that this mutation causes predominantly cryptic splicing, with a 7-nucleotide deletion in exon 12. The ensuing frame shift disrupts the last 54 amino acids of plakophilin-2 and extends the open reading frame by 145 nucleotides (48 amino acids) into the 3' untranslated region. Haplotype analysis demonstrates the absence of remote consanguinity. Heterozygous family members produce approximately 60% of properly spliced PKP2 and do not have manifestations of ARVD. Further analysis of PKP2 mRNA sequence revealed two additional alternatively spliced transcripts. The possibility of cryptic or alternative splicing should be considered with identification of apparently synonymous nucleotide substitutions in this gene.
(c) 2006 Wiley-Liss, Inc.
Figures
References
-
- Alcalai R, Metzger S, Rosenheck S, Meiner V, Chajek-Shaul T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J Am Coll Cardiol. 2003;42(2):319–327. - PubMed
-
- Bauce B, Basso C, Rampazzo A, Beffagna G, Daliento L, Frigo G, Malacrida S, Settimo L, Danieli G, Thiene G, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26(16):1666–1675. - PubMed
-
- Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65(2):366–373. - PubMed
-
- Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. 2002;277(12):10512–10522. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
