

X-linked Opitz G/BBB syndrome: identification of a novel mutation and prenatal diagnosis in a Korean family
- PMID: 17043407
- PMCID: PMC2721984
- DOI: 10.3346/jkms.2006.21.5.790
X-linked Opitz G/BBB syndrome: identification of a novel mutation and prenatal diagnosis in a Korean family
Abstract
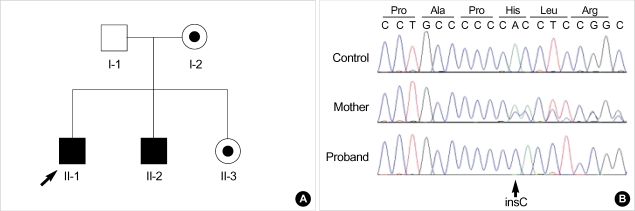
X-linked Opitz G/BBB syndrome (XLOS; MIM 300000) is a rare multiple congenital anomaly disorder that is characterized by facial anomalies, laryngeal/tracheal/esophageal defects and genitourinary abnormalities. XLOS is caused by mutations in the MID1 gene which encodes a microtubule-associated RING-Bbox-Coiled-coil (RBCC) protein. We recently found a four-year Korean male patient who was suspected of having XLOS. Mutation analysis of the MID1 gene in the patient and his mother demonstrated that the patient had a novel insertion mutation (c.1798_1799-insC), and his mother was a heterozygous carrier of the mutation. After identification of the causative mutation in this family, prenatal diagnosis of two consecutive fetuses were successfully undertaken. This is the first report on a genetically confirmed case of XLOS in Korea.
Figures
Similar articles
-
Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations.Am J Med Genet A. 2005 Jan 1;132A(1):1-7. doi: 10.1002/ajmg.a.30407. Am J Med Genet A. 2005. PMID: 15558842
-
A novel mutation in MID1 in a patient with X-linked Opitz G/BBB syndrome.Gene. 2014 Mar 1;537(1):140-2. doi: 10.1016/j.gene.2013.12.018. Epub 2013 Dec 26. Gene. 2014. PMID: 24374473
-
Complex rearrangement of the exon 6 genomic region among Opitz G/BBB Syndrome MID1 alterations.Eur J Med Genet. 2013 Aug;56(8):404-10. doi: 10.1016/j.ejmg.2013.05.009. Epub 2013 Jun 19. Eur J Med Genet. 2013. PMID: 23791568
-
X-linked Opitz syndrome: novel mutations in the MID1 gene and redefinition of the clinical spectrum.Am J Med Genet A. 2003 Jul 15;120A(2):222-8. doi: 10.1002/ajmg.a.10265. Am J Med Genet A. 2003. PMID: 12833403 Review.
-
First trimester ultrasound features of X-linked Opitz syndrome and early molecular diagnosis: case report and review of the literature.J Matern Fetal Neonatal Med. 2021 Sep;34(18):3089-3093. doi: 10.1080/14767058.2019.1677594. Epub 2019 Oct 21. J Matern Fetal Neonatal Med. 2021. PMID: 31630581 Review.
Cited by
-
Two Novel Pathogenic MID1 Variants and Genotype-Phenotype Correlation Reanalysis in X-Linked Opitz G/BBB Syndrome.Mol Syndromol. 2017 Dec;9(1):45-51. doi: 10.1159/000479177. Epub 2017 Aug 29. Mol Syndromol. 2017. PMID: 29456483 Free PMC article.
-
Mid1/Mid2 expression in craniofacial development and a literature review of X-linked opitz syndrome.Mol Genet Genomic Med. 2015 Dec 12;4(1):95-105. doi: 10.1002/mgg3.183. eCollection 2016 Jan. Mol Genet Genomic Med. 2015. PMID: 26788540 Free PMC article.
-
Exome sequencing improves genetic diagnosis of congenital orofacial clefts.Front Genet. 2023 Sep 7;14:1252823. doi: 10.3389/fgene.2023.1252823. eCollection 2023. Front Genet. 2023. PMID: 37745857 Free PMC article.
-
Opitz GBBB syndrome with total anomalous pulmonary venous connection: A new MID1 gene variant.Mol Genet Genomic Med. 2023 Sep;11(9):e2234. doi: 10.1002/mgg3.2234. Epub 2023 Jul 27. Mol Genet Genomic Med. 2023. PMID: 37498300 Free PMC article.
-
Auditory findings and electrophysiologics in individuals with G/BBB syndrome.Braz J Otorhinolaryngol. 2011 Nov-Dec;77(6):768-74. doi: 10.1590/S1808-86942011000600014. Braz J Otorhinolaryngol. 2011. PMID: 22183284 Free PMC article.
References
-
- Opitz JM, Guttenberger JE, Pellet JR. The G syndrome of multiple congenital anomalies. Birth Defects Orig Artic Ser (V) 1969;2:95–102.
-
- Opitz JM, Smith DW. The BBB syndrome. Familial telecanthus with associated congenital anomalies. Birth Defects Orig Artic Ser (V) 1969;2:86–94.
-
- Robin NH, Opitz JM, Muenke M. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am J Med Genet. 1996;62:305–317. - PubMed
-
- Robin NH, Feldman GJ, Aronson AL, Mitchell HF, Weksberg R, Leonard CO, Burton BK, Josephson KD, Laxova R, Aleck KA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat Genet. 1995;11:459–461. - PubMed
-
- Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet. 1997;17:285–291. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
