

Pathway and gene-set activation measurement from mRNA expression data: the tissue distribution of human pathways
- PMID: 17044931
- PMCID: PMC1794557
- DOI: 10.1186/gb-2006-7-10-r93
Pathway and gene-set activation measurement from mRNA expression data: the tissue distribution of human pathways
Abstract
Background: Interpretation of lists of genes or proteins with altered expression is a critical and time-consuming part of microarray and proteomics research, but relatively little attention has been paid to methods for extracting biological meaning from these output lists. One powerful approach is to examine the expression of predefined biological pathways and gene sets, such as metabolic and signaling pathways and macromolecular complexes. Although many methods for measuring pathway expression have been proposed, a systematic analysis of the performance of multiple methods over multiple independent data sets has not previously been reported.
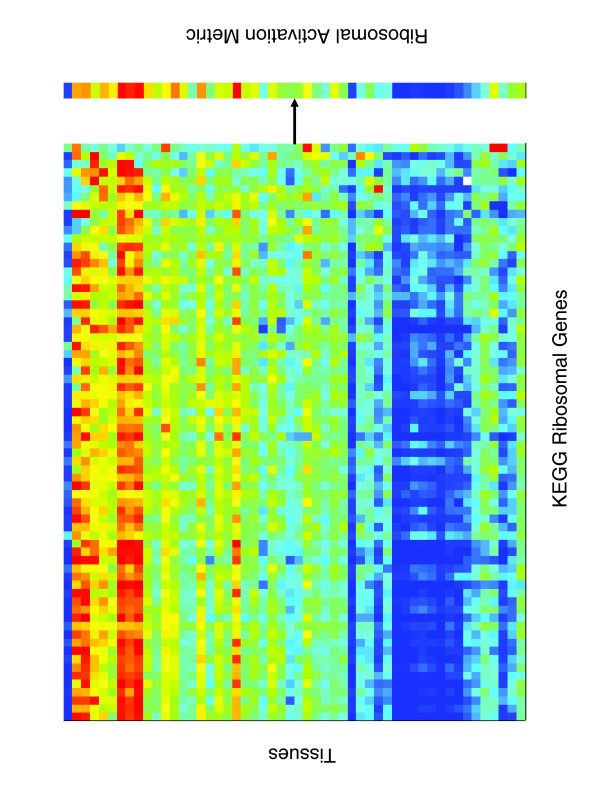
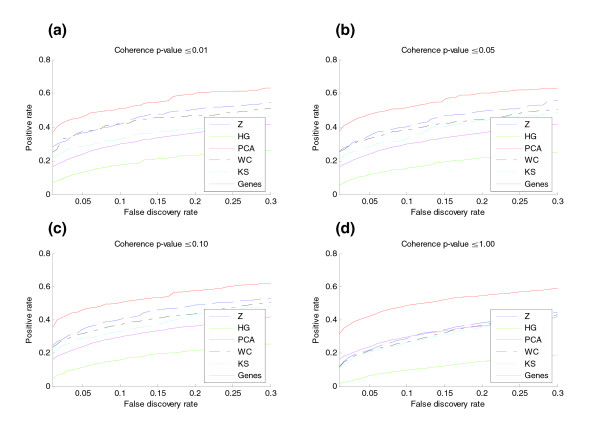
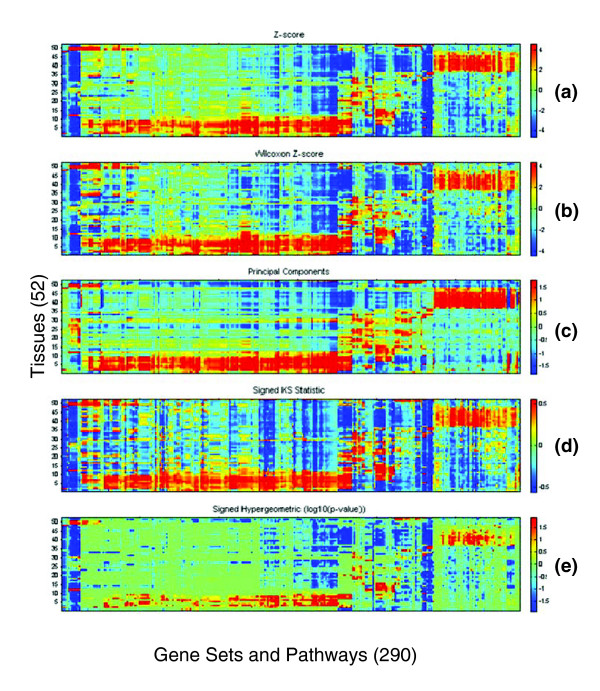
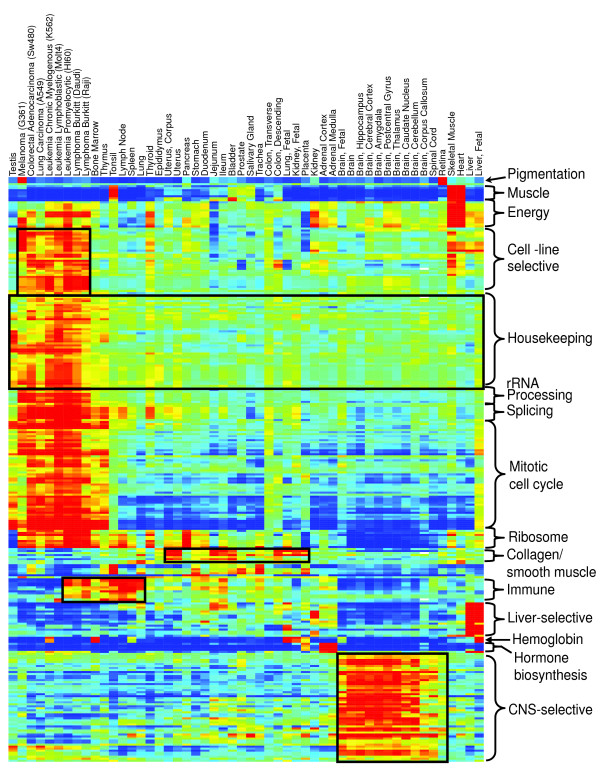
Results: Five different measures of pathway expression were compared in an analysis of nine publicly available mRNA expression data sets. The relative sensitivity of the metrics varied greatly across data sets, and the biological pathways identified for each data set are also dependent on the choice of pathway activation metric. In addition, we show that removing incoherent pathways prior to analysis improves specificity. Finally, we create and analyze a public map of pathway expression in human tissues by gene-set analysis of a large compendium of human expression data.
Conclusion: We show that both the detection sensitivity and identity of pathways significantly perturbed in a microarray experiment are highly dependent on the analysis methods used and how incoherent pathways are treated. Analysts should thus consider using multiple approaches to test the robustness of their biological interpretations. We also provide a comprehensive picture of the tissue distribution of human gene pathways and a useful public archive of human pathway expression data.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
