

Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection
- PMID: 17053089
- PMCID: PMC1665635
- DOI: 10.1101/gr.5655606
Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection
Abstract
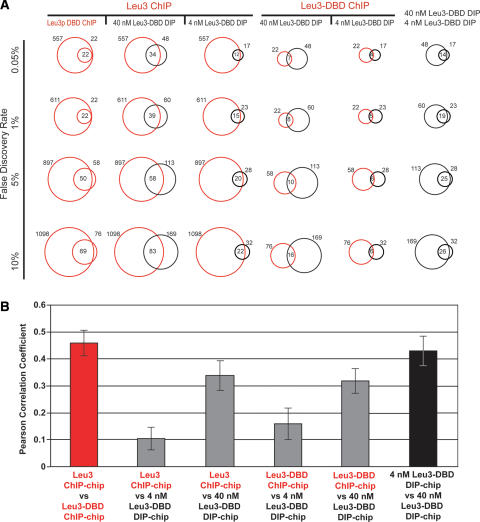
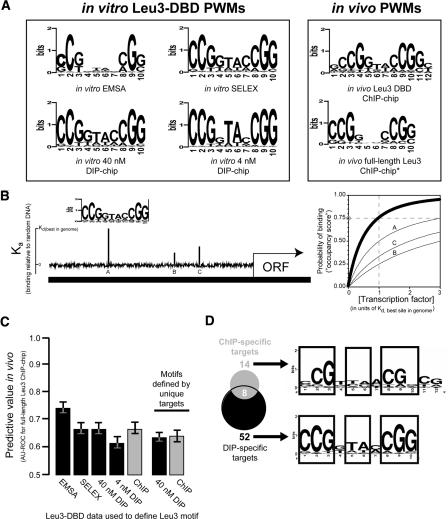
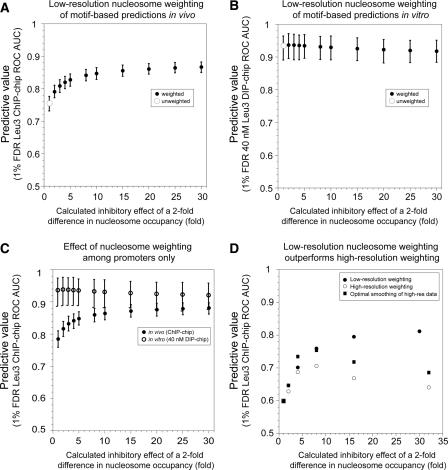
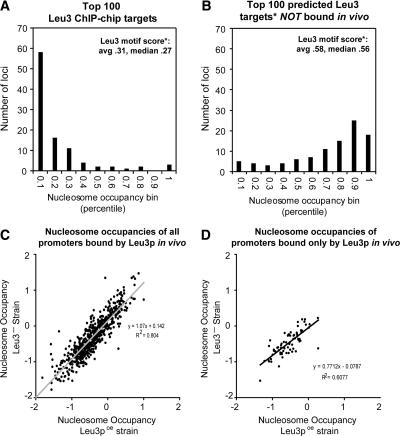
Sequence motifs that are potentially recognized by DNA-binding proteins occur far more often in genomic DNA than do observed in vivo protein-DNA interactions. To determine how chromatin influences the utilization of particular DNA-binding sites, we compared the in vivo genome-wide binding location of the yeast transcription factor Leu3 to the binding location observed on the same genomic DNA in the absence of any protein cofactors. We found that the DNA-sequence motif recognized by Leu3 in vitro and in vivo was functionally indistinguishable, but Leu3 bound different genomic locations under the two conditions. Accounting for nucleosome occupancy in addition to DNA-sequence motifs significantly improved the prediction of protein-DNA interactions in vivo, but not the prediction of sites bound by purified Leu3 in vitro. Use of histone modification data does not further improve binding predictions, presumably because their effect is already manifest in the global histone distribution. Measurements of nucleosome occupancy in strains that differ in Leu3 genotype show that low nucleosome occupancy at loci bound by Leu3 is not a consequence of Leu3 binding. These results permit quantitation of the epigenetic influence that chromatin exerts on DNA binding-site selection, and provide evidence for an instructive, functionally important role for nucleosome occupancy in determining patterns of regulatory factor targeting genome-wide.
Figures
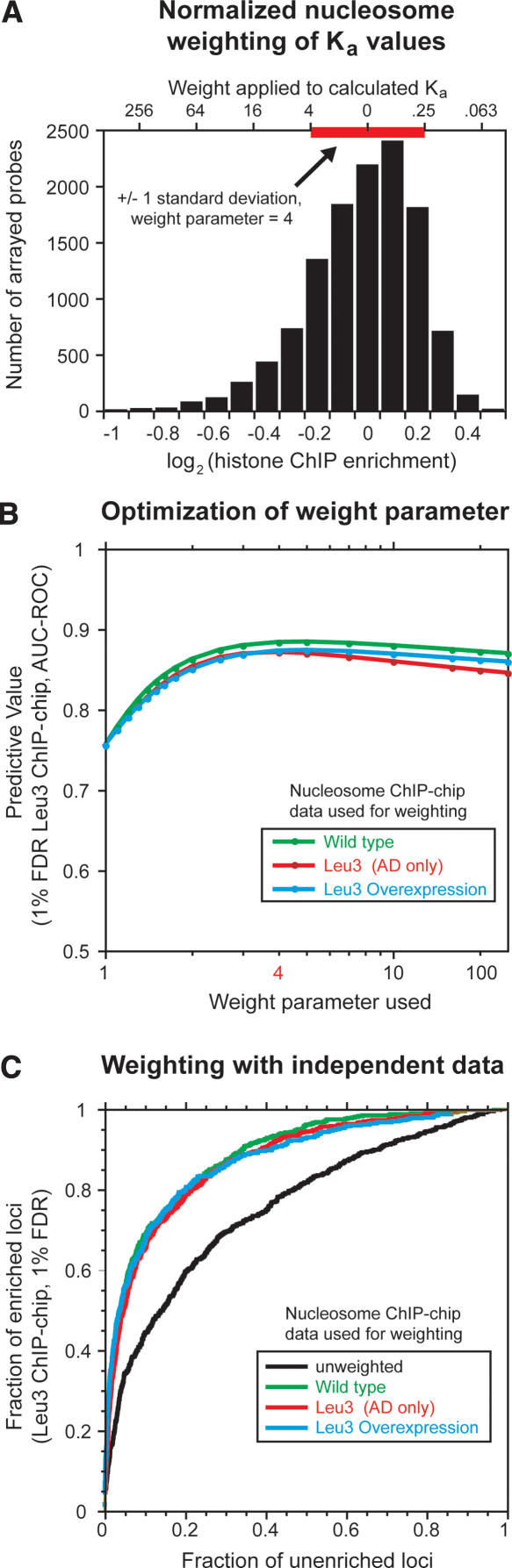
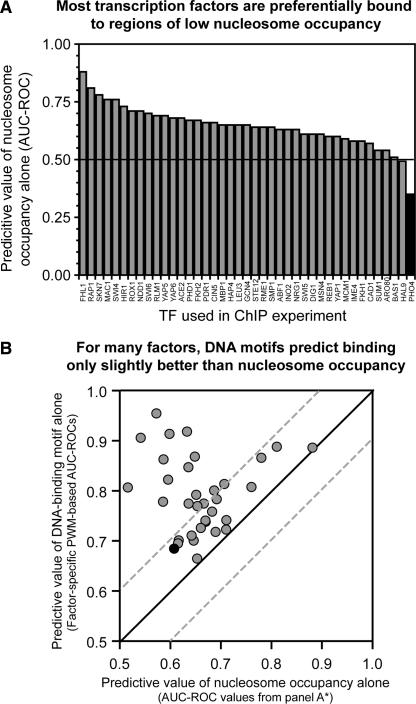
References
-
- Brachmann C.B., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D., Davies A., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D., Cost G.J., Caputo E., Li J., Hieter P., Boeke J.D., Caputo E., Li J., Hieter P., Boeke J.D., Li J., Hieter P., Boeke J.D., Hieter P., Boeke J.D., Boeke J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. - PubMed
-
- Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Belle A., Dephoure N., O’Shea E.K., Weissman J.S., Dephoure N., O’Shea E.K., Weissman J.S., O’Shea E.K., Weissman J.S., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases