

Towards a conceptual and operational union of bacterial systematics, ecology, and evolution
- PMID: 17062416
- PMCID: PMC1764936
- DOI: 10.1098/rstb.2006.1918
Towards a conceptual and operational union of bacterial systematics, ecology, and evolution
Abstract
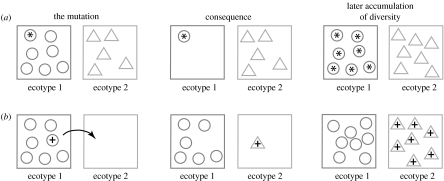
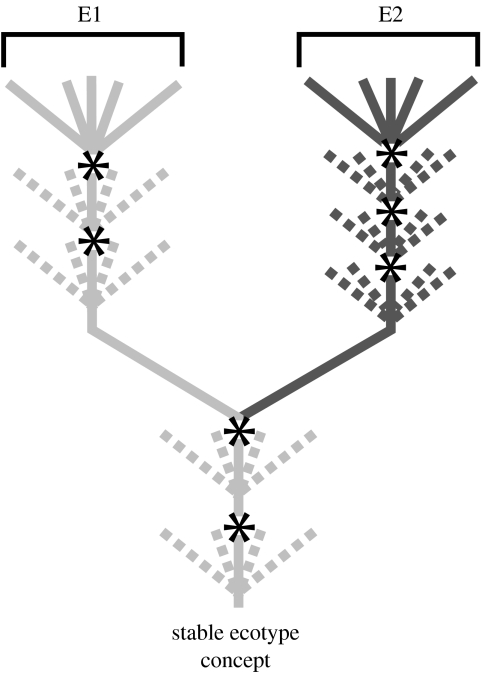
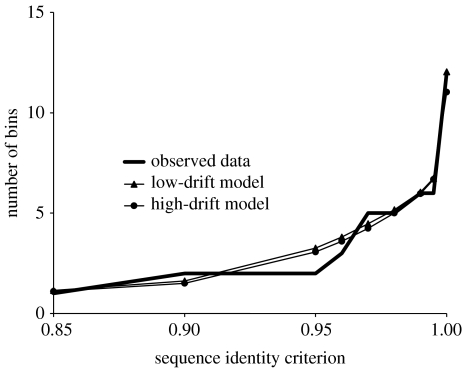
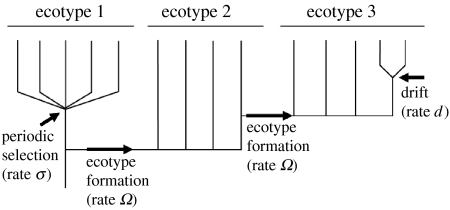
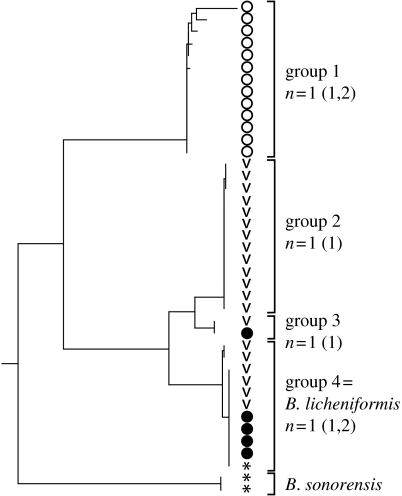
To completely understand the ecology of a bacterial community, we need to identify its ecologically distinct populations (ecotypes). The greatest promise for enumerating a community's constituent ecotypes is held by molecular approaches that identify bacterial ecotypes as DNA sequence clusters. These approaches succeed when ecotypes correspond with sequence clusters, but some models of bacterial speciation predict a one-to-many and others a many-to-one relationship between ecotypes and sequence clusters. A further challenge is that sequence-based phylogenies often contain a hierarchy of clusters and subclusters within clusters, and there is no widely accepted theory to guide systematists and ecologists to the size of cluster most likely to correspond to ecotypes. While present systematics attempts to use universal thresholds of sequence divergence to help demarcate species, the recently developed 'community phylogeny' approach assumes no universal thresholds, but demarcates ecotypes based on the analysis of a lineage's evolutionary dynamics. Theory-based approaches like this one can give a conceptual framework as well as operational criteria for hypothesizing the identity and membership of ecotypes from sequence data; ecology-based approaches can then confirm that the putative ecotypes are actually ecologically distinct. Bacterial ecotypes that are demonstrated to have a history of coexistence as ecologically distinct lineages (based on sequence analysis) and as a prognosis of future coexistence (based on ecological differences), are the fundamental units of bacterial ecology and evolution, and should be recognized by bacterial systematics.
Figures
References
-
- Acinas S.G, Klepac-Ceraj V, Hunt D.E, Pharino C, Ceraj I, Distel D.L, Polz M.F. Fine-scale phylogenetic architecture of a complex bacterial community. Nature. 2004;430:551–554. doi:10.1038/nature02649 - DOI - PubMed
-
- Alm R.A, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi:10.1038/16495 - DOI - PubMed
-
- Baas-Becking L.G.M. The Hague. W.P. Van Stockum & Zoon; The Netherlands: 1934. Geobiologie of Inleiding tot de Milieukunde.
-
- Bansal A.K, Meyer T.E. Evolutionary analysis by whole-genome comparisons. J. Bacteriol. 2002;184:2260–2272. doi:10.1128/JB.184.8.2260-2272.2002 - DOI - PMC - PubMed
-
- Beijerinck M.W. Müller; Amsterdam, The Netherlands: 1913. Jaarboek van de Koninklijke Akademie v. Wetenschappen.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources