

Combinatorial methods for small-molecule placement in computational enzyme design
- PMID: 17075051
- PMCID: PMC1636520
- DOI: 10.1073/pnas.0607691103
Combinatorial methods for small-molecule placement in computational enzyme design
Abstract
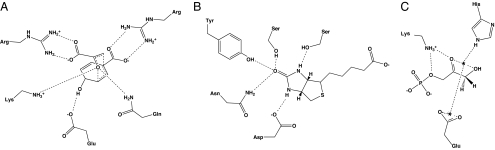
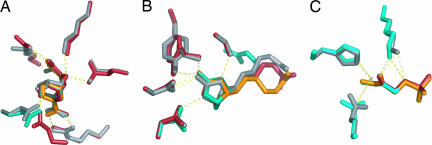
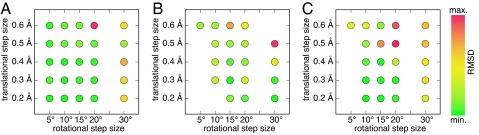
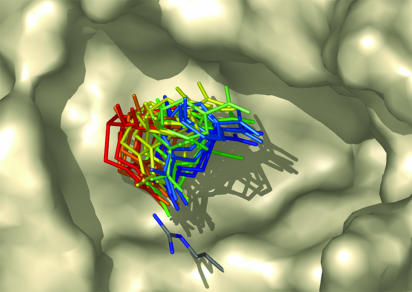
The incorporation of small-molecule transition state structures into protein design calculations poses special challenges because of the need to represent the added translational, rotational, and conformational freedoms within an already difficult optimization problem. Successful approaches to computational enzyme design have focused on catalytic side-chain contacts to guide placement of small molecules in active sites. We describe a process for modeling small molecules in enzyme design calculations that extends previously described methods, allowing favorable small-molecule positions and conformations to be explored simultaneously with sequence optimization. Because all current computational enzyme design methods rely heavily on sampling of possible active site geometries from discrete conformational states, we tested the effects of discretization parameters on calculation results. Rotational and translational step sizes as well as side-chain library types were varied in a series of computational tests designed to identify native-like binding contacts in three natural systems. We find that conformational parameters, especially the type of rotamer library used, significantly affect the ability of design calculations to recover native binding-site geometries. We describe the construction and use of a crystallographic conformer library and find that it more reliably captures active-site geometries than traditional rotamer libraries in the systems tested.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
