

A backbone-based theory of protein folding
- PMID: 17075053
- PMCID: PMC1636505
- DOI: 10.1073/pnas.0606843103
A backbone-based theory of protein folding
Abstract
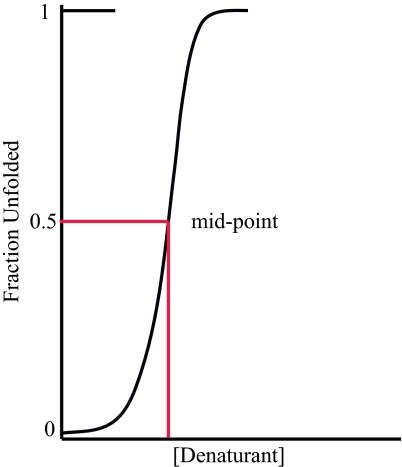
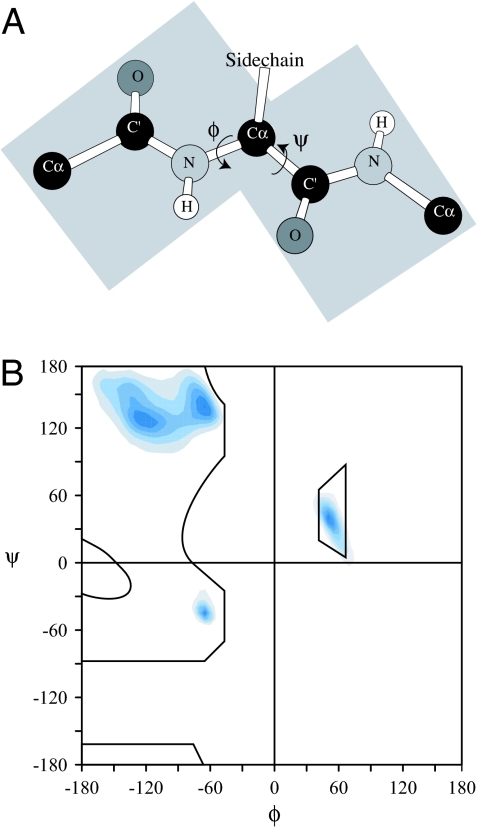
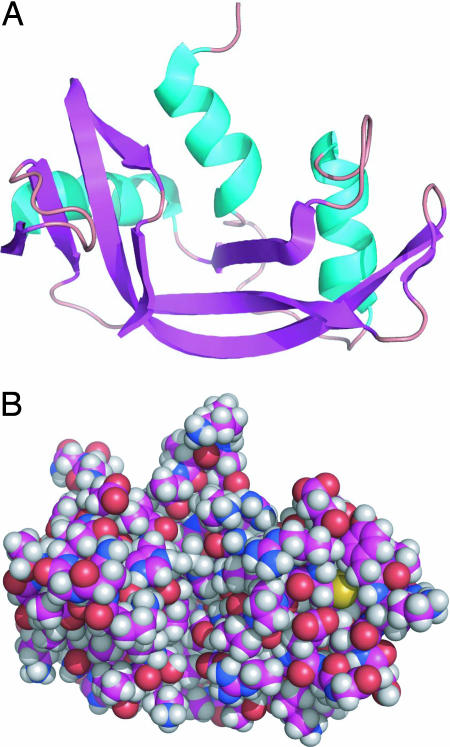
Under physiological conditions, a protein undergoes a spontaneous disorder order transition called "folding." The protein polymer is highly flexible when unfolded but adopts its unique native, three-dimensional structure when folded. Current experimental knowledge comes primarily from thermodynamic measurements in solution or the structures of individual molecules, elucidated by either x-ray crystallography or NMR spectroscopy. From the former, we know the enthalpy, entropy, and free energy differences between the folded and unfolded forms of hundreds of proteins under a variety of solvent/cosolvent conditions. From the latter, we know the structures of approximately 35,000 proteins, which are built on scaffolds of hydrogen-bonded structural elements, alpha-helix and beta-sheet. Anfinsen showed that the amino acid sequence alone is sufficient to determine a protein's structure, but the molecular mechanism responsible for self-assembly remains an open question, probably the most fundamental open question in biochemistry. This perspective is a hybrid: partly review, partly proposal. First, we summarize key ideas regarding protein folding developed over the past half-century and culminating in the current mindset. In this view, the energetics of side-chain interactions dominate the folding process, driving the chain to self-organize under folding conditions. Next, having taken stock, we propose an alternative model that inverts the prevailing side-chain/backbone paradigm. Here, the energetics of backbone hydrogen bonds dominate the folding process, with preorganization in the unfolded state. Then, under folding conditions, the resultant fold is selected from a limited repertoire of structural possibilities, each corresponding to a distinct hydrogen-bonded arrangement of alpha-helices and/or strands of beta-sheet.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
