

Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans
- PMID: 17078022
- PMCID: PMC4144349
- DOI: 10.1002/humu.20429
Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans
Abstract
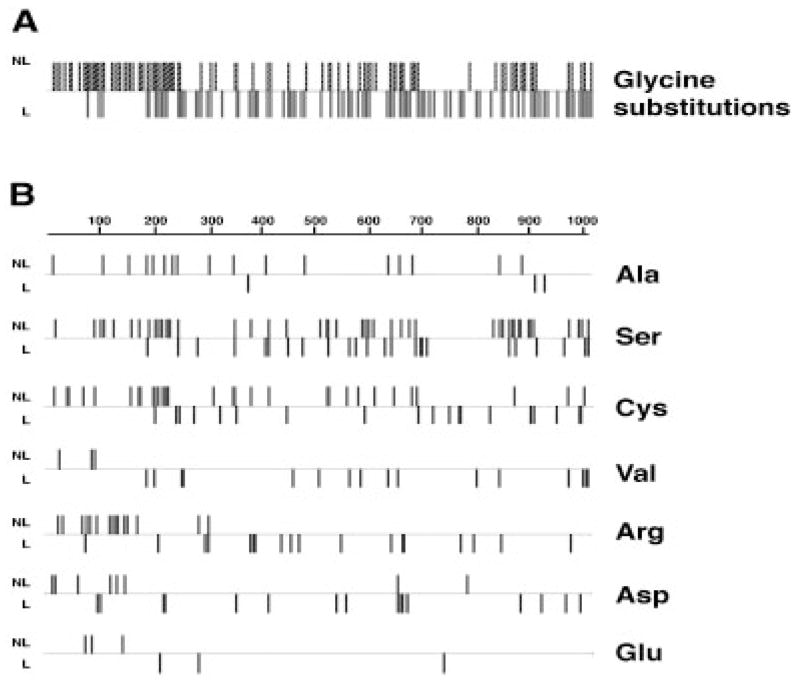
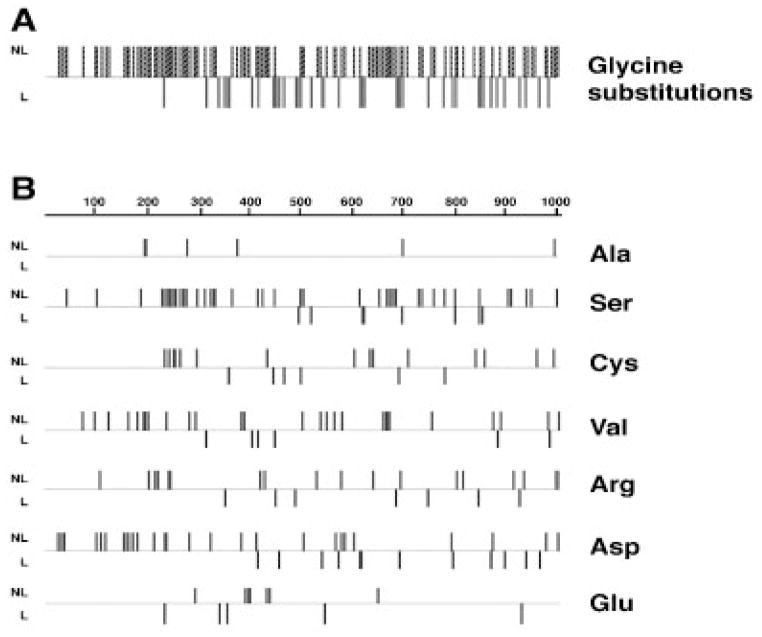
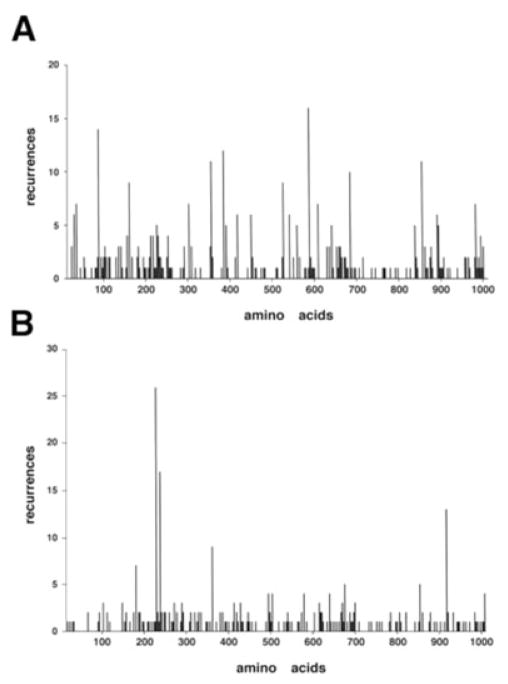
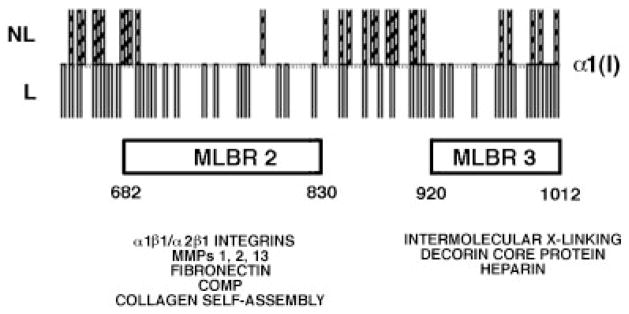
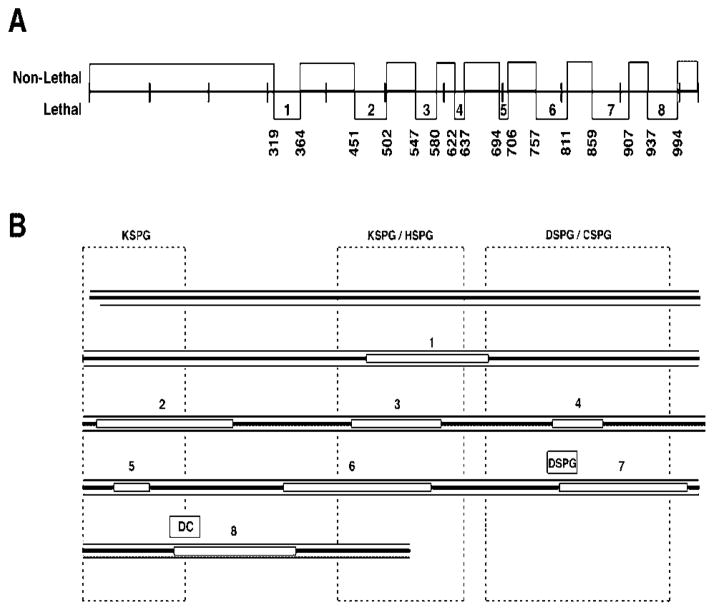
Osteogenesis imperfecta (OI) is a generalized disorder of connective tissue characterized by fragile bones and easy susceptibility to fracture. Most cases of OI are caused by mutations in type I collagen. We have identified and assembled structural mutations in type I collagen genes (COL1A1 and COL1A2, encoding the proalpha1(I) and proalpha2(I) chains, respectively) that result in OI. Quantitative defects causing type I OI were not included. Of these 832 independent mutations, 682 result in substitution for glycine residues in the triple helical domain of the encoded protein and 150 alter splice sites. Distinct genotype-phenotype relationships emerge for each chain. One-third of the mutations that result in glycine substitutions in alpha1(I) are lethal, especially when the substituting residues are charged or have a branched side chain. Substitutions in the first 200 residues are nonlethal and have variable outcome thereafter, unrelated to folding or helix stability domains. Two exclusively lethal regions (helix positions 691-823 and 910-964) align with major ligand binding regions (MLBRs), suggesting crucial interactions of collagen monomers or fibrils with integrins, matrix metalloproteinases (MMPs), fibronectin, and cartilage oligomeric matrix protein (COMP). Mutations in COL1A2 are predominantly nonlethal (80%). Lethal substitutions are located in eight regularly spaced clusters along the chain, supporting a regional model. The lethal regions align with proteoglycan binding sites along the fibril, suggesting a role in fibril-matrix interactions. Recurrences at the same site in alpha2(I) are generally concordant for outcome, unlike alpha1(I). Splice site mutations comprise 20% of helical mutations identified in OI patients, and may lead to exon skipping, intron inclusion, or the activation of cryptic splice sites. Splice site mutations in COL1A1 are rarely lethal; they often lead to frameshifts and the mild type I phenotype. In alpha2(I), lethal exon skipping events are located in the carboxyl half of the chain. Our data on genotype-phenotype relationships indicate that the two collagen chains play very different roles in matrix integrity and that phenotype depends on intracellular and extracellular events.
Figures
References
-
- Ayad S, Boot-Handford RP, Humphries MS, Kadler CA. The extracellular matrix facts book. London: Academic Press; 1998.
-
- Bächinger HP, Davis JM. Sequence specific thermal stability of the collagen triple helix. Int J Biol Macromol. 1991;13:152–156. - PubMed
-
- Bächinger HP, Morris NP, Davis JM. Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the helix of type I collagen. Am J Med Genet. 1993;45:152–162. - PubMed
-
- Barnes AM, Chang W, Morello R, Cabral WA, Weis MA, Eyre DR, Makareeva E, Kouznetsova N, Leikin S, Uveges TE, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC. Recessive lethal form of osteogenesis imperfecta caused by null mutations in CRTAP. 28th Annual Meeting of American Society of Bone and Mineral Research; Philadelphia, PA. 2006. p. Abstract 922.
-
- Baronas-Lowell D, Lauer-Fields JL, Fields GB. Induction of endothelial cell activation by a triple helical α2β1 integrin ligand, derived from type I collagen α1(I)496–507. J Biol Chem. 2004;279:952–962. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
