

Dysfunction of TGF-beta signaling in Alzheimer's disease
- PMID: 17080189
- PMCID: PMC1626134
- DOI: 10.1172/JCI30284
Dysfunction of TGF-beta signaling in Alzheimer's disease
Abstract
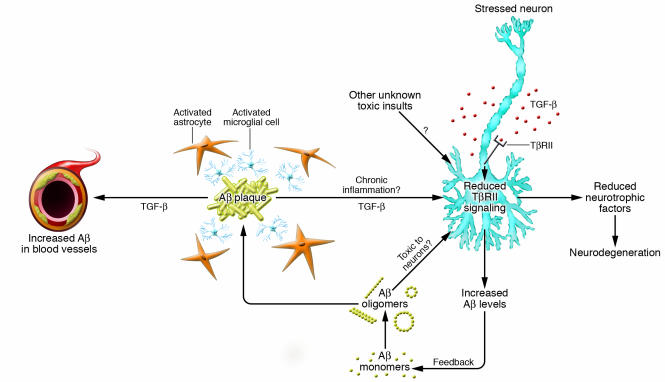
Accumulation of beta-amyloid peptide (Abeta) in the brain is believed to trigger a complex and poorly understood pathologic reaction that results in the development of Alzheimer's disease (AD). Despite intensive study, there is no consensus as to how Abeta accumulation causes neurodegeneration in AD. In this issue of the JCI, Tesseur et al. report that the expression of TGF-beta type II receptor (TbetaRII) by neurons is reduced very early in the course of AD and that reduced TGF-beta signaling increased Abeta deposition and neurodegeneration in a mouse model of AD (see the related article beginning on page 3060). Intriguingly, reduced TGF-beta signaling in neuroblastoma cells resulted in neuritic dystrophy and increased levels of secreted Abeta. Collectively, these data suggest that dysfunction of the TGF-beta/TbetaRII signaling axis in the AD brain may accelerate Abeta deposition and neurodegeneration.
Figures
Comment on
-
Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology.J Clin Invest. 2006 Nov;116(11):3060-9. doi: 10.1172/JCI27341. J Clin Invest. 2006. PMID: 17080199 Free PMC article.
References
-
- Wyss-Coray T., Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002;35:419–432. - PubMed
-
- Glabe C.C. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell. Biochem. 2005;38:167–177. - PubMed
-
- Kayed R., et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. - PubMed
-
- Walsh D.M., et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
