

Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology
- PMID: 17080199
- PMCID: PMC1626127
- DOI: 10.1172/JCI27341
Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology
Abstract
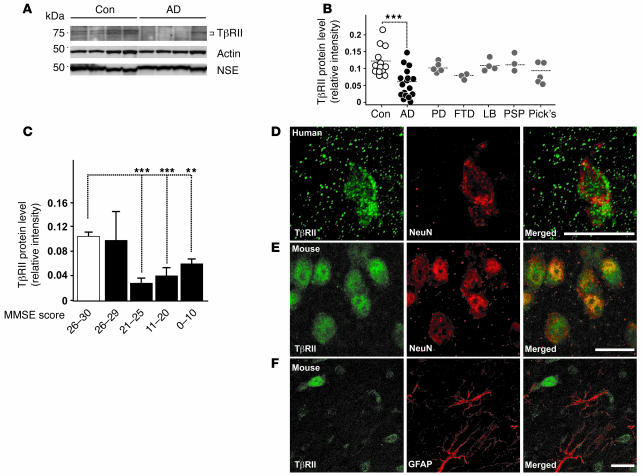
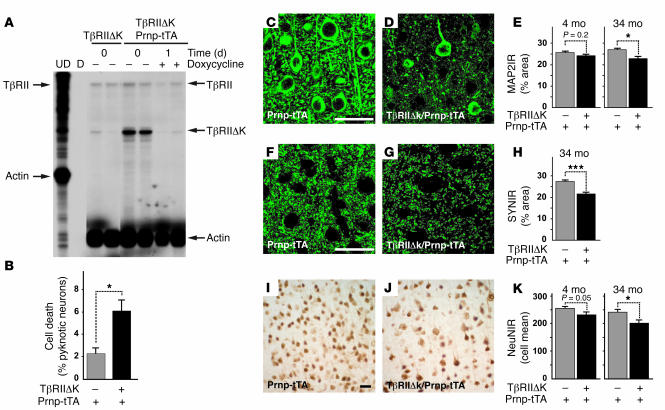
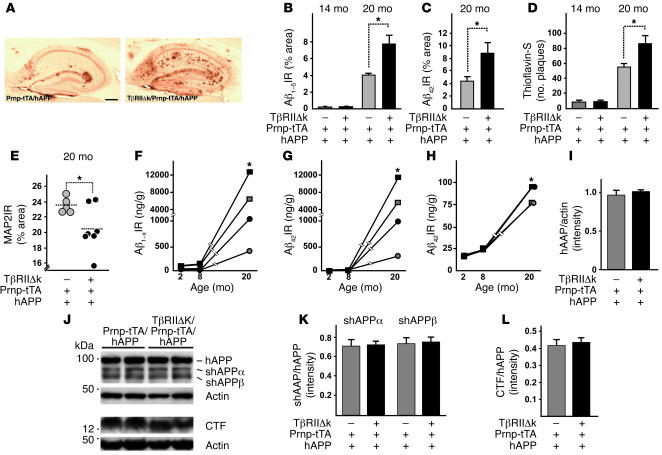
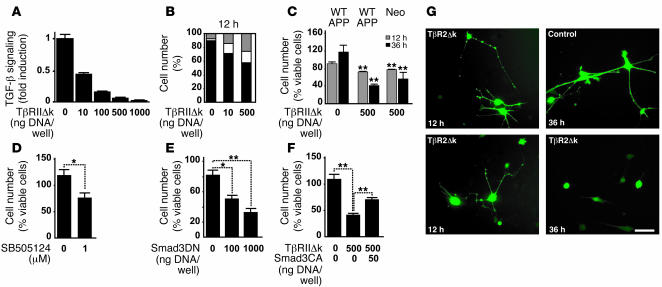
Alzheimer's disease (AD) is characterized by progressive neurodegeneration and cerebral accumulation of the beta-amyloid peptide (Abeta), but it is unknown what makes neurons susceptible to degeneration. We report that the TGF-beta type II receptor (TbetaRII) is mainly expressed by neurons, and that TbetaRII levels are reduced in human AD brain and correlate with pathological hallmarks of the disease. Reducing neuronal TGF-beta signaling in mice resulted in age-dependent neurodegeneration and promoted Abeta accumulation and dendritic loss in a mouse model of AD. In cultured cells, reduced TGF-beta signaling caused neuronal degeneration and resulted in increased levels of secreted Abeta and beta-secretase-cleaved soluble amyloid precursor protein. These results show that reduced neuronal TGF-beta signaling increases age-dependent neurodegeneration and AD-like disease in vivo. Increasing neuronal TGF-beta signaling may thus reduce neurodegeneration and be beneficial in AD.
Figures
Comment in
-
Dysfunction of TGF-beta signaling in Alzheimer's disease.J Clin Invest. 2006 Nov;116(11):2855-7. doi: 10.1172/JCI30284. J Clin Invest. 2006. PMID: 17080189 Free PMC article.
References
-
- Selkoe D.J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. - PubMed
-
- Connor B., Dragunow M. The role of neuronal growth factors in neurodegenerative disorders of the human brain. Brain Res. Brain Res. Rev. 1998;27:1–39. - PubMed
-
- Murer M.G., Yan Q., Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog. Neurobiol. 2001;63:71–124. - PubMed
-
- Allen S.J., Wilcock G.K., Dawbarn D. Profound and selective loss of catalytic TrkB immunoreactivity in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1999;264:648–651. - PubMed
-
- Ferrer I., et al. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999;58:729–739. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
