

Molecular dynamics simulations of RNA: an in silico single molecule approach
- PMID: 17080418
- PMCID: PMC2018183
- DOI: 10.1002/bip.20620
Molecular dynamics simulations of RNA: an in silico single molecule approach
Abstract
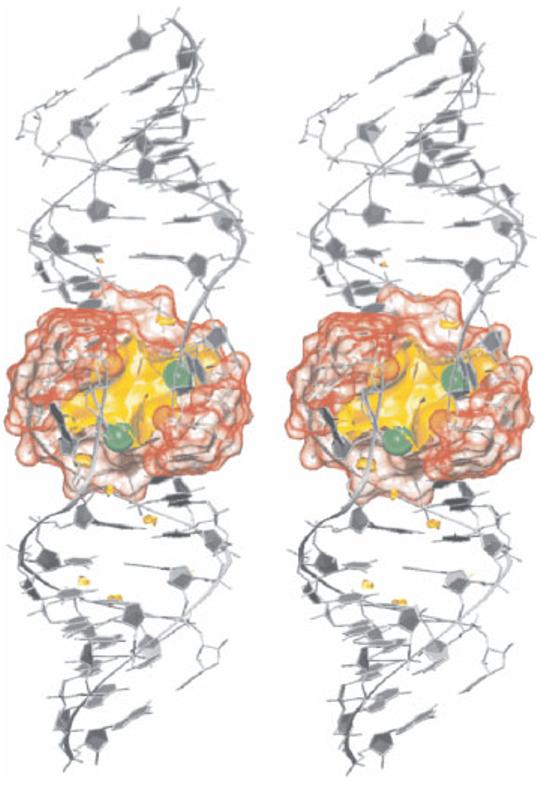
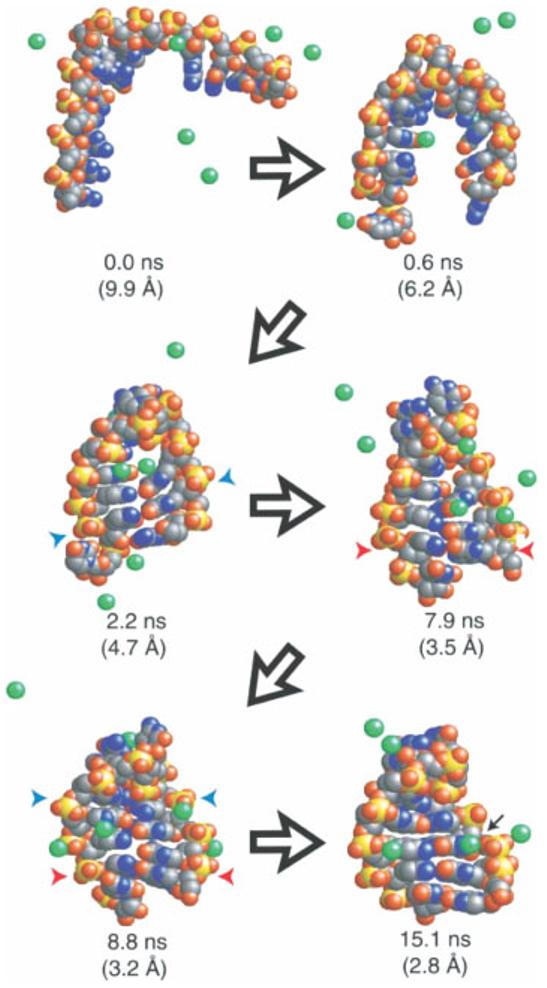
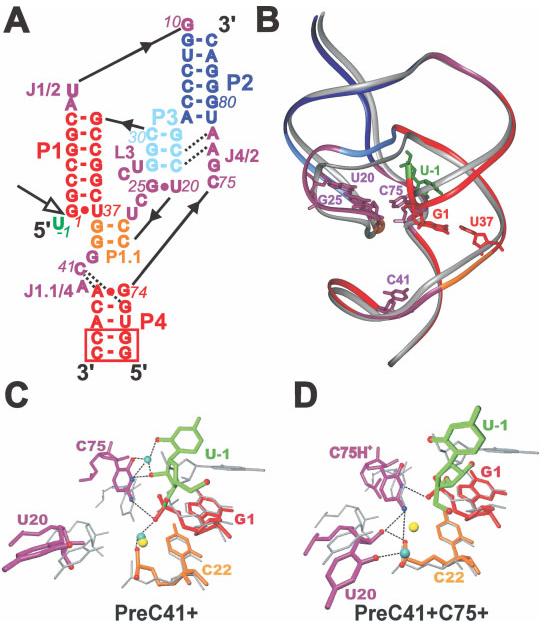
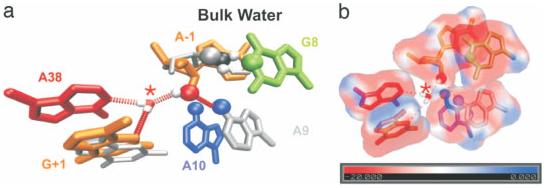
RNA molecules are now known to be involved in the processing of genetic information at all levels, taking on a wide variety of central roles in the cell. Understanding how RNA molecules carry out their biological functions will require an understanding of structure and dynamics at the atomistic level, which can be significantly improved by combining computational simulation with experiment. This review provides a critical survey of the state of molecular dynamics (MD) simulations of RNA, including a discussion of important current limitations of the technique and examples of its successful application. Several types of simulations are discussed in detail, including those of structured RNA molecules and their interactions with the surrounding solvent and ions, catalytic RNAs, and RNA-small molecule and RNA-protein complexes. Increased cooperation between theorists and experimentalists will allow expanded judicious use of MD simulations to complement conceptually related single molecule experiments. Such cooperation will open the door to a fundamental understanding of the structure-function relationships in diverse and complex RNA molecules. .
(c) 2006 Wiley Periodicals, Inc.
Figures
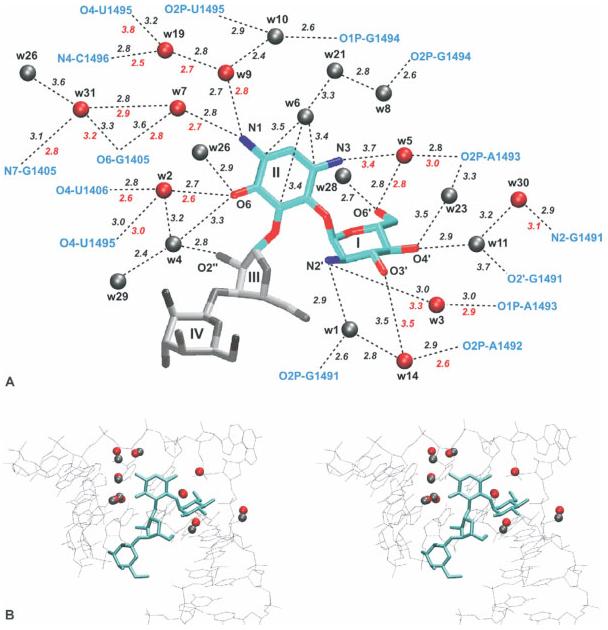
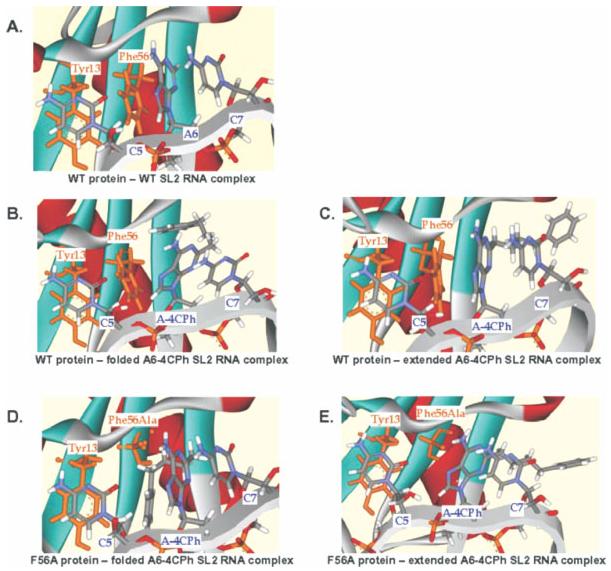
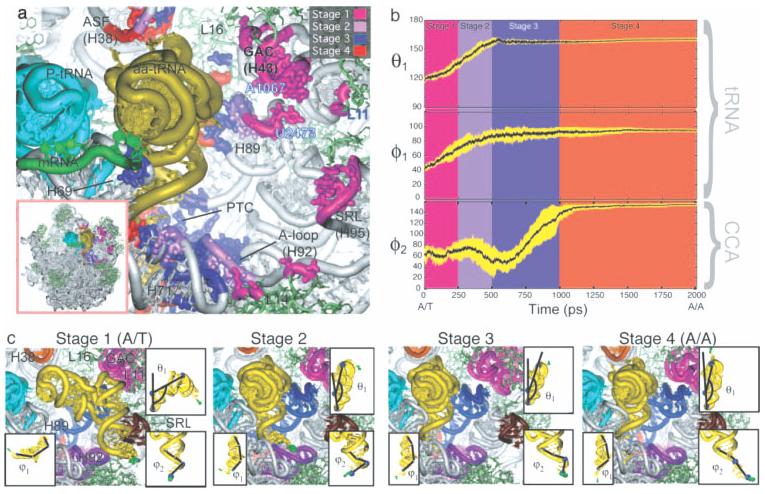
References
-
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Cell. 1982;31:147–158. - PubMed
-
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. Cell. 1983;35:849–857. - PubMed
-
- Flores R, Delgado S, Gas ME, Carbonell A, Molina D, Gago S, De la Pena M. FEBS Lett. 2004;567:42–48. - PubMed
-
- Dorsett Y, Tuschl T. Nat Rev Drug Discov. 2004;3:318–329. - PubMed
-
- Hannon GJ, Rossi JJ. Nature. 2004;431:371–378. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
