

A statistical score for assessing the quality of multiple sequence alignments
- PMID: 17081313
- PMCID: PMC1687212
- DOI: 10.1186/1471-2105-7-484
A statistical score for assessing the quality of multiple sequence alignments
Abstract
Background: Multiple sequence alignment is the foundation of many important applications in bioinformatics that aim at detecting functionally important regions, predicting protein structures, building phylogenetic trees etc. Although the automatic construction of a multiple sequence alignment for a set of remotely related sequences cause a very challenging and error-prone task, many downstream analyses still rely heavily on the accuracy of the alignments.
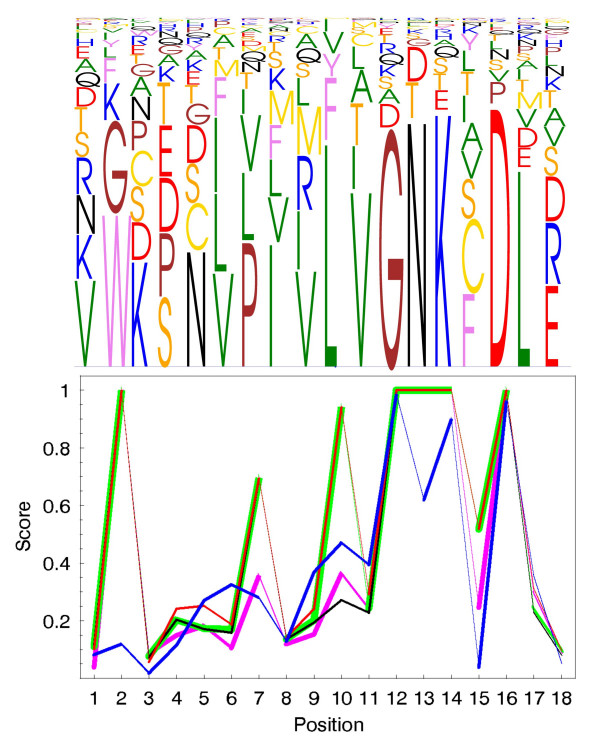
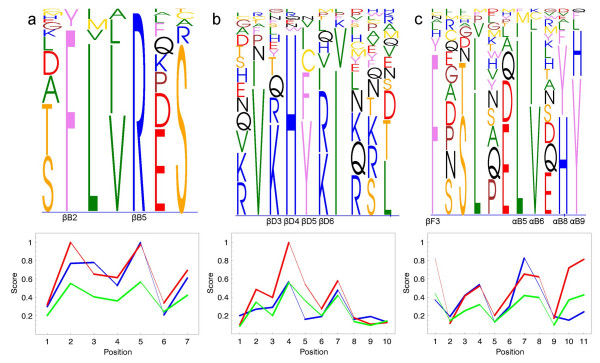
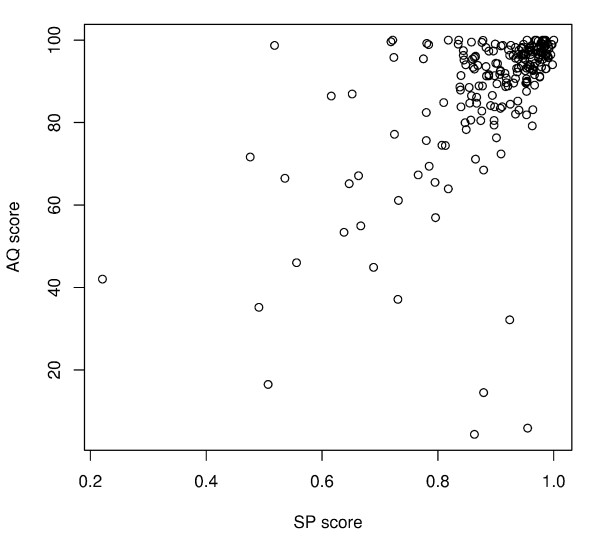
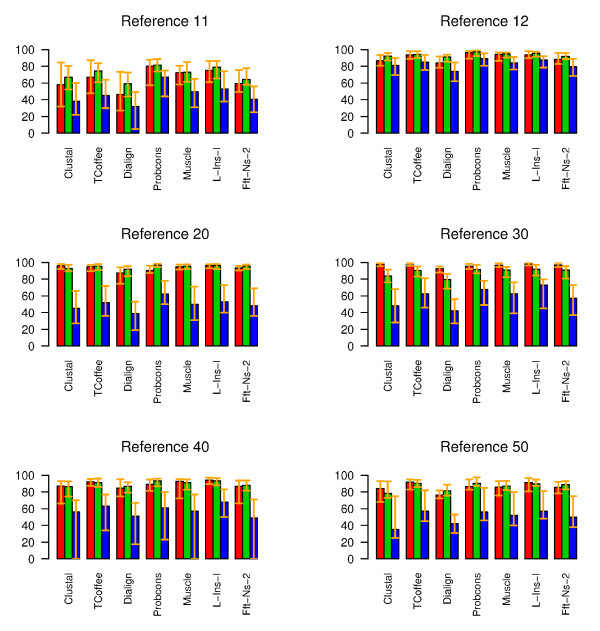
Results: To address the need for an objective evaluation framework, we introduce a statistical score that assesses the quality of a given multiple sequence alignment. The quality assessment is based on counting the number of significantly conserved positions in the alignment using importance sampling method in conjunction with statistical profile analysis framework. We first evaluate a novel objective function used in the alignment quality score for measuring the positional conservation. The results for the Src homology 2 (SH2) domain, Ras-like proteins, peptidase M13, subtilase and beta-lactamase families demonstrate that the score can distinguish sequence patterns with different degrees of conservation. Secondly, we evaluate the quality of the alignments produced by several widely used multiple sequence alignment programs using a novel alignment quality score and a commonly used sum of pairs method. According to these results, the Mafft strategy L-INS-i outperforms the other methods, although the difference between the Probcons, TCoffee and Muscle is mostly insignificant. The novel alignment quality score provides similar results than the sum of pairs method.
Conclusion: The results indicate that the proposed statistical score is useful in assessing the quality of multiple sequence alignments.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
