

A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase
- PMID: 17081990
- PMCID: PMC1855097
- DOI: 10.1016/j.molcel.2006.09.015
A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase
Abstract
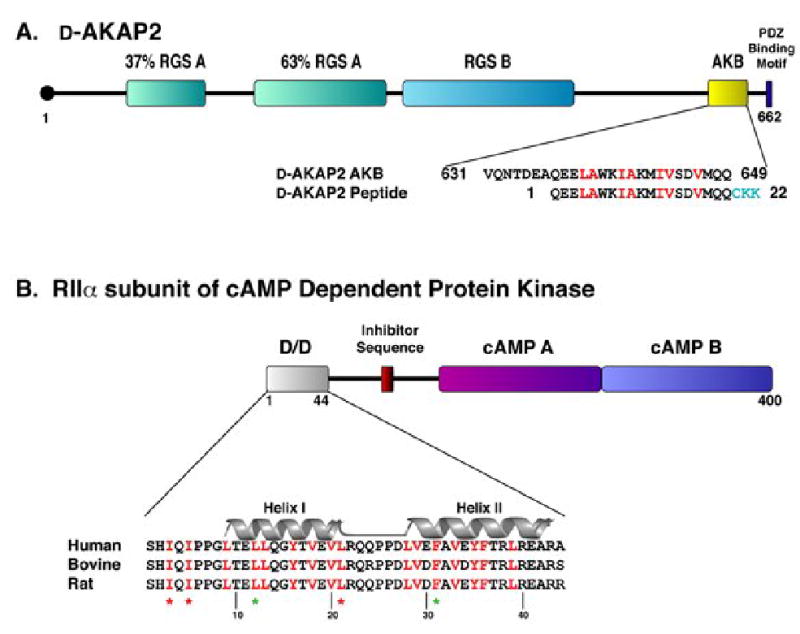
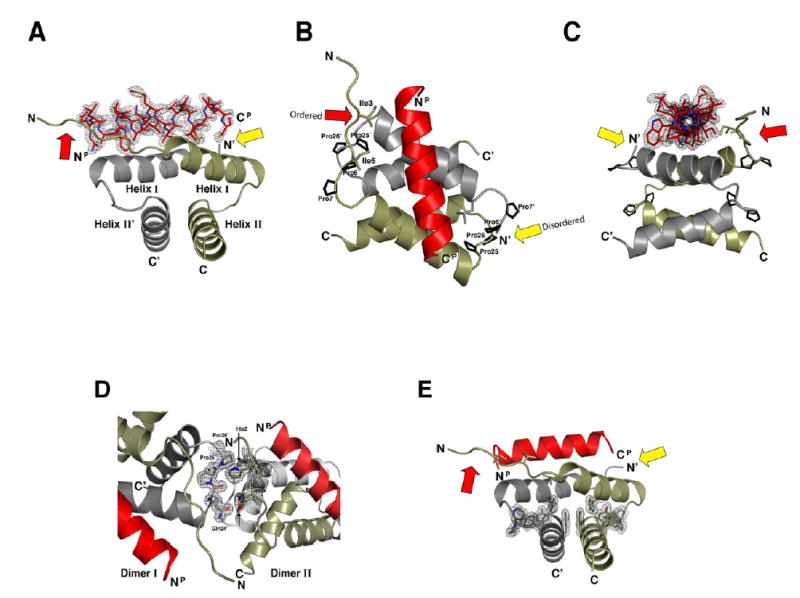
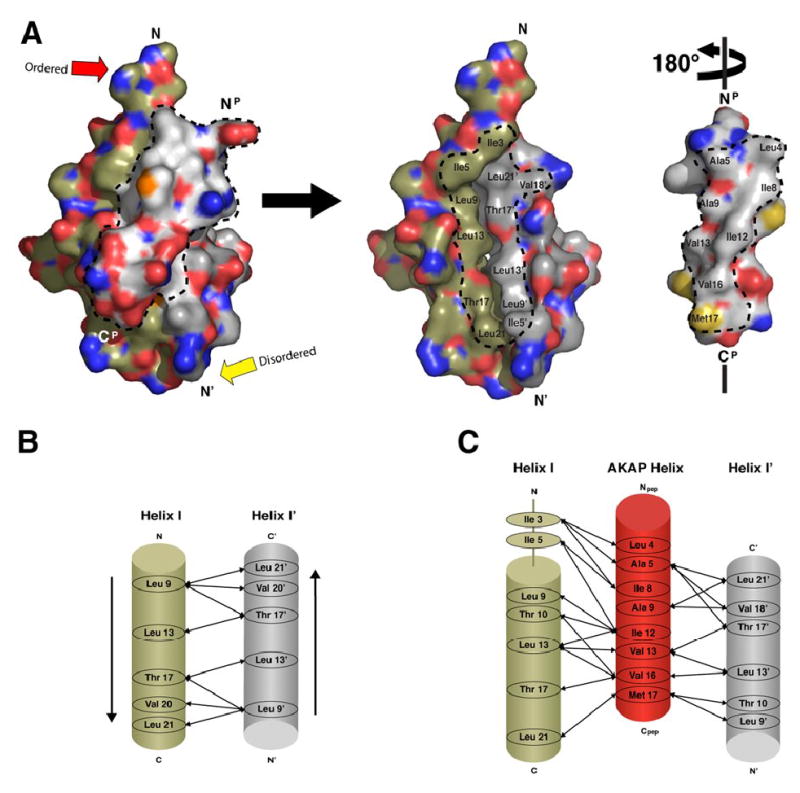
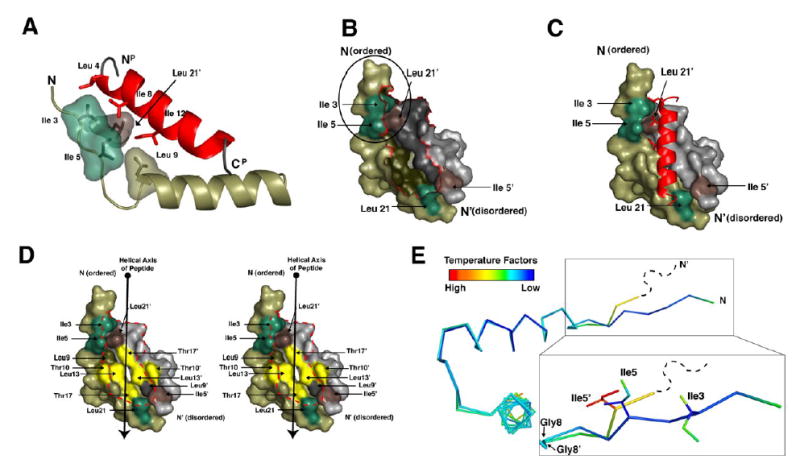
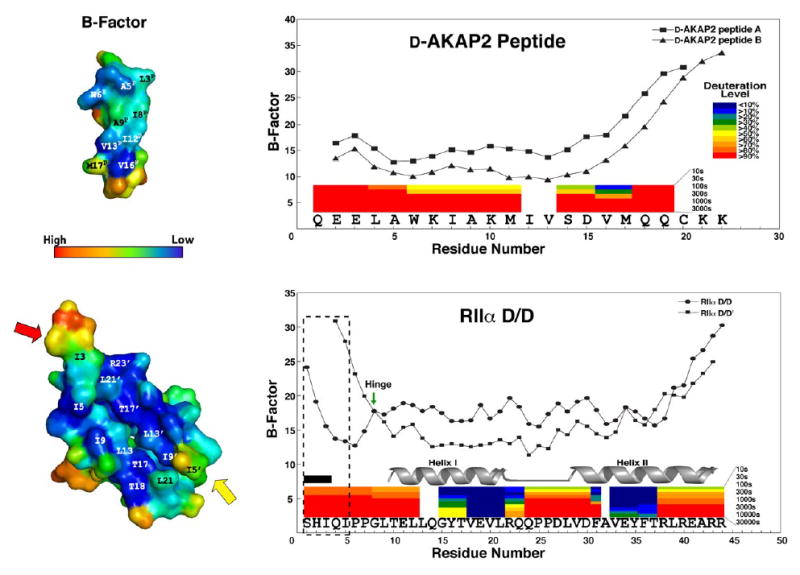
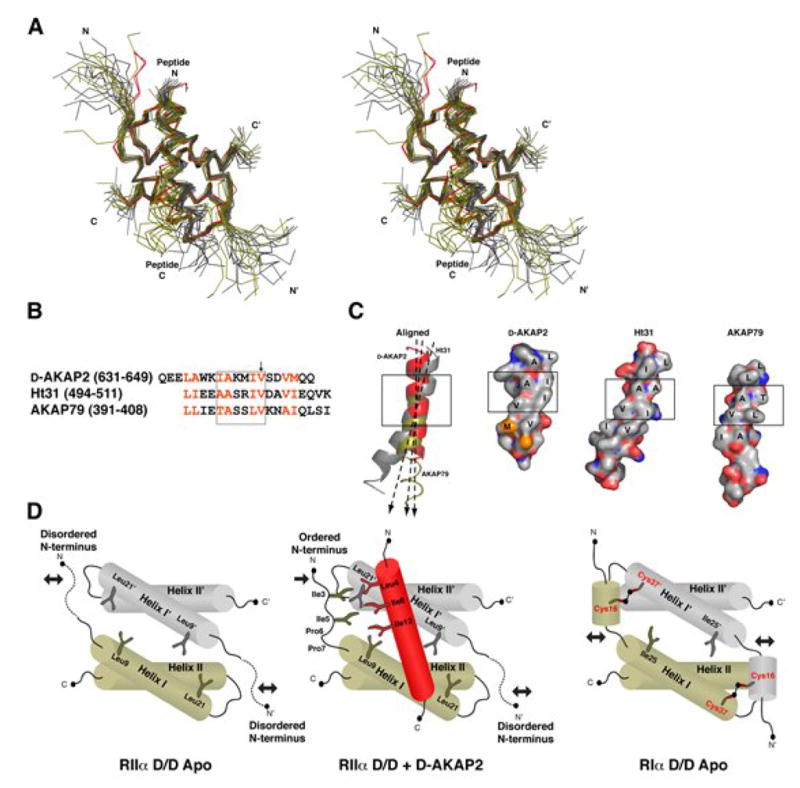
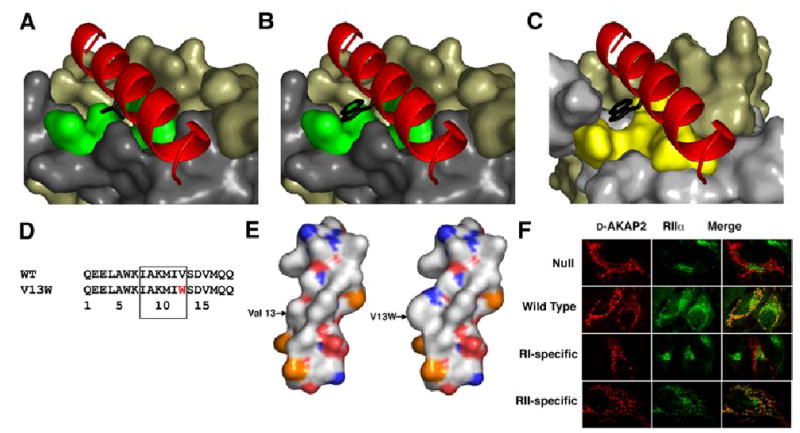
A kinase-anchoring proteins (AKAPs) target PKA to specific microdomains by using an amphipathic helix that docks to N-terminal dimerization and docking (D/D) domains of PKA regulatory (R) subunits. To understand specificity, we solved the crystal structure of the helical motif from D-AKAP2, a dual-specific AKAP, bound to the RIIalpha D/D domain. The 1.6 Angstrom structure reveals how this dynamic, hydrophobic docking site is assembled. A stable, hydrophobic docking groove is formed by the helical interface of two RIIalpha protomers. The flexible N terminus of one protomer is then recruited to the site, anchored to the peptide through two essential isoleucines. The other N terminus is disordered. This asymmetry provides greater possibilities for AKAP docking. Although there is strong discrimination against RIalpha in the N terminus of the AKAP helix, the hydrophobic groove discriminates against RIIalpha. RIalpha, with a cavity in the groove, can accept a bulky tryptophan, whereas RIIalpha requires valine.
Figures
References
-
- Banky P, Roy M, Newlon MG, Morikis D, Haste NM, Taylor SS, Jennings PA. Related protein-protein interaction modules present drastically different surface topographies despite a conserved helical platform. J Mol Biol. 2003;330:1117–1129. - PubMed
-
- Burns LL, Canaves JM, Pennypacker JK, Blumenthal DK, Taylor SS. Isoform specific differences in binding of a dual-specificity A-kinase anchoring protein to type I and type II regulatory subunits of PKA. Biochemistry. 2003;42:5754–5763. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
