

Cell biology of polycystin-2
- PMID: 17084592
- PMCID: PMC1817723
- DOI: 10.1016/j.cellsig.2006.09.005
Cell biology of polycystin-2
Abstract
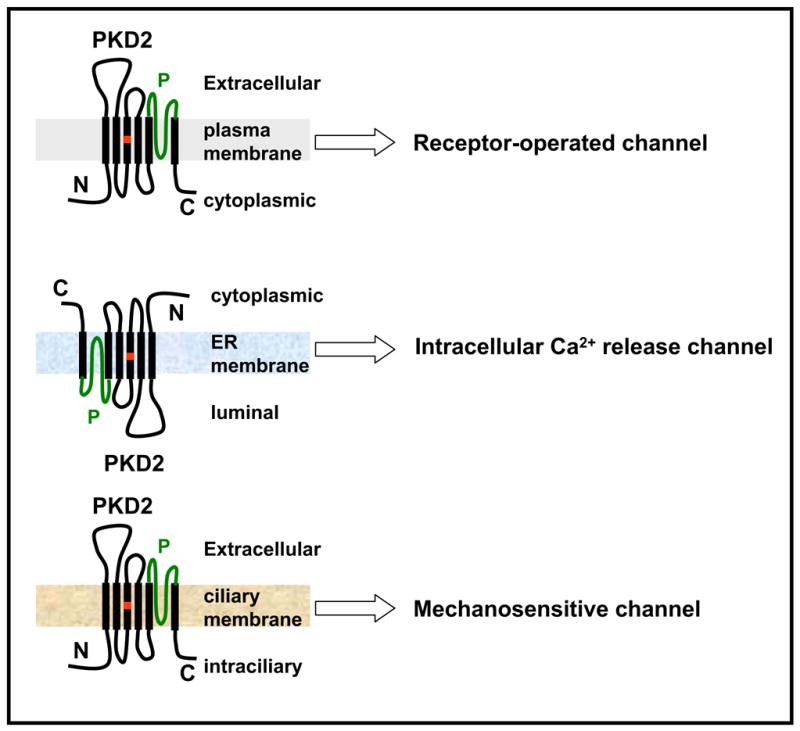
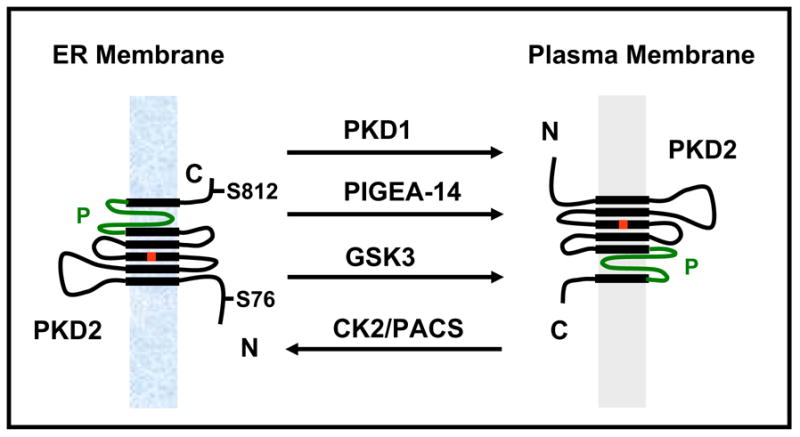
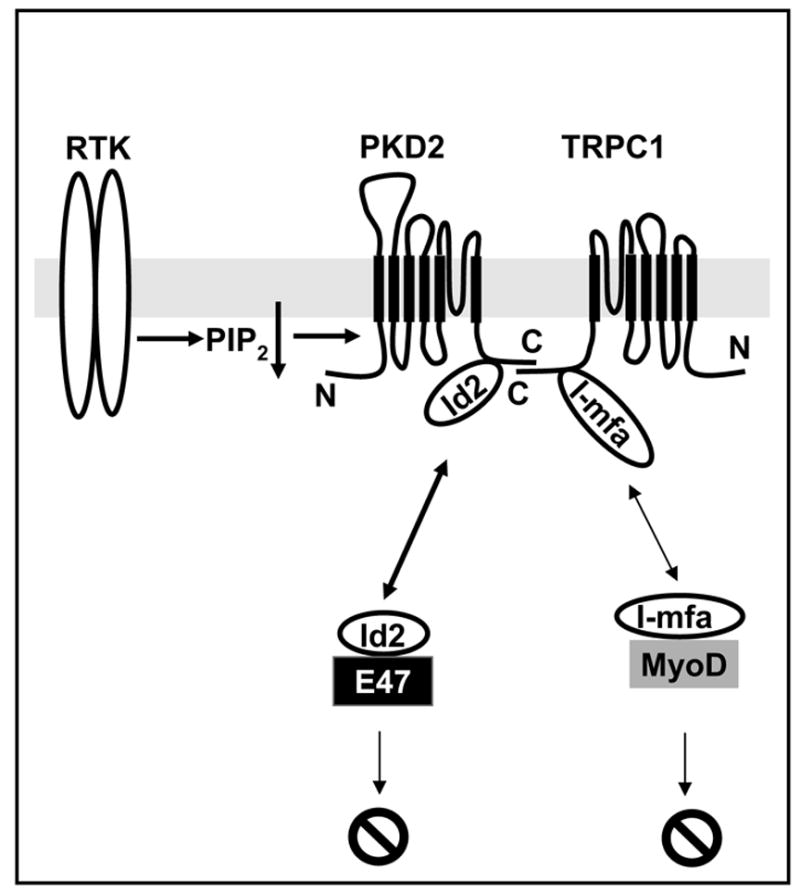
Naturally occurring mutations in two separate, but interacting loci, pkd1 and pkd2 are responsible for almost all cases of autosomal dominant polycystic kidney disease (ADPKD). ADPKD is one of the most common genetic diseases resulting primarily in the formation of large kidney, liver, and pancreatic cysts. Homozygous deletion of either pkd1 or pkd2 results in embryonic lethality in mice due to kidney and heart defects illustrating their indispensable roles in mammalian development. However, the mechanism by which mutations in these genes cause ADPKD and other developmental defects are unknown. Research in the past several years has revealed that PKD2 has multiple functions depending on its subcellular localization. It forms a receptor-operated, non-selective cation channel in the plasma membrane, a novel intracellular Ca2+ release channel in the endoplasmic reticulum (ER), and a mechanosensitive channel in the primary cilium. This review focuses on the functional compartmentalization of PKD2, its modes of activation, and PKD2-mediated signal transduction.
Figures
References
-
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. Science. 1996;272(5266):1339–1342. - PubMed
-
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. Nat Genet. 1995;10(2):151–160. - PubMed
-
- Consortium EPKD. Cell. 1994;77(6):881–894. - PubMed
-
- Consortium IPKD. Cell. 1995;81(2):289–298. - PubMed
-
- Gabow PA. N Engl J Med. 1993;329(5):332–342. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
