

Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide
- PMID: 17088865
- PMCID: PMC2014634
- DOI: 10.1038/sj.bjp.0706953
Lovastatin protects human endothelial cells from the genotoxic and cytotoxic effects of the anticancer drugs doxorubicin and etoposide
Abstract
Background and purpose: 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statins) are frequently used lipid-lowering drugs. Moreover, they exert pleiotropic effects on cellular stress responses and death. Here, we analysed whether lovastatin affects the sensitivity of primary human endothelial cells (HUVEC) to the anticancer drug doxorubicin.
Experimental approach: We investigated whether pretreatment of HUVEC with low dose of lovastatin influences the cellular sensitivity to doxorubicin. To this end, cell viability, proliferation and apoptosis as well as DNA damage-triggered stress response were analysed.
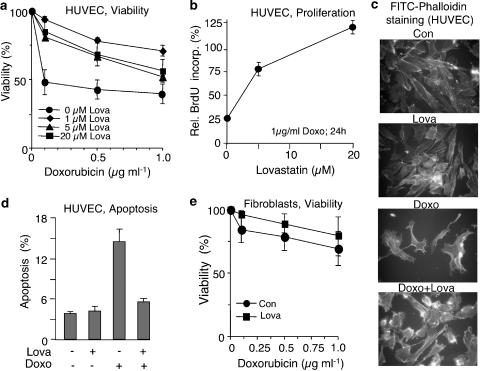
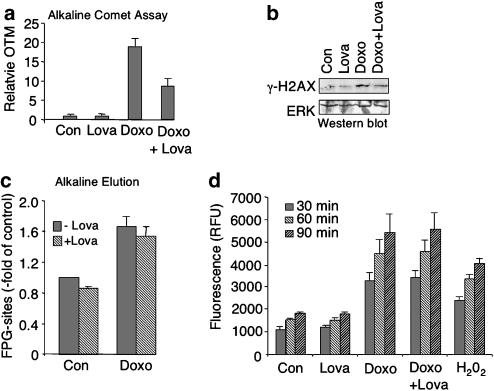
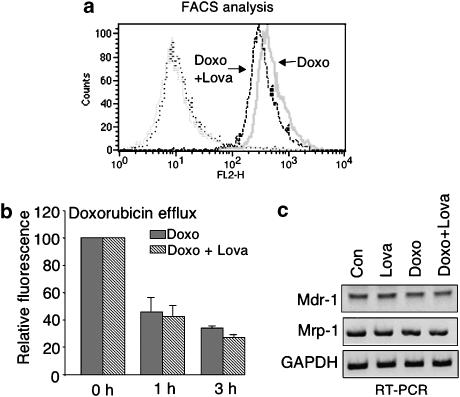
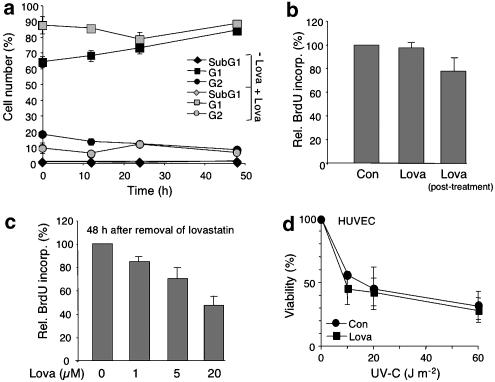
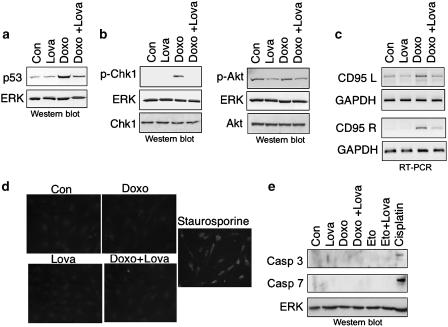
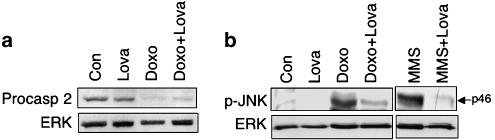
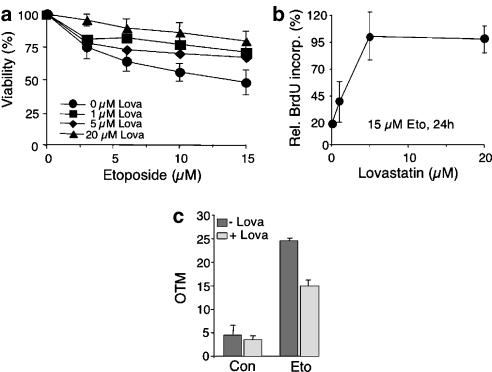
Key results: Lovastatin reduced the cytotoxic potency of doxorubicin in HUVEC. Lovastatin attenuated the doxorubicin-induced increase in p53 as well as activation of checkpoint kinase (Chk-1) and stress-activated protein kinase/c-Jun-N-terminal kinase (SAPK/JNK). Acquired doxorubicin resistance was independent of alterations in doxorubicin efflux and cell cycle progression. Also, doxorubicin-triggered production of reactive oxygen species (ROS) and formation of oxidative DNA lesions remained unaffected by lovastatin. However, lovastatin impaired DNA strand break formation induced by doxorubicin. Notably, lovastatin also conferred cross-resistance to the cytotoxic and genotoxic effects of etoposide, indicating that lovastatin shields topoisomerase II against poisons.
Conclusions and implications: Based on these data, we suggest that lovastatin-mediated resistance to topoisomerase II inhibitors is due to a reduction in DNA damage and, hence, it attenuates stress responses leading to cell death that are triggered by DNA damage. Therefore, lovastatin might be useful clinically for alleviating side-effects of anticancer therapies that include topoisomerase II inhibitors.
Figures
References
-
- Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. - PubMed
-
- Bachur NR, Yu F, Johnson R, Hickey R, Wu Y, Malkas L. Helicase inhibition by anthracycline anticancer agents. Mol Pharmacol. 1992;41:993–998. - PubMed
-
- Brown GA, McPherson JP, Gu L, Hedley DW, Toso R, Deuchars KL, et al. Relationship of DNA topoisomerase II alpha and beta expression to cytotoxicity of antineoplastic agents in human acute lymphoblastic leukemia cell lines. Cancer Res. 1995;55:78–82. - PubMed
-
- Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26:883–891. - PubMed
-
- Canman CE, Kastan MB. Signal transduction. Three paths to stress relief. Nature. 1996;384:213–214. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
