

Disease-associated mutations at copper ligand histidine residues of superoxide dismutase 1 diminish the binding of copper and compromise dimer stability
- PMID: 17092942
- PMCID: PMC2757151
- DOI: 10.1074/jbc.M604503200
Disease-associated mutations at copper ligand histidine residues of superoxide dismutase 1 diminish the binding of copper and compromise dimer stability
Abstract
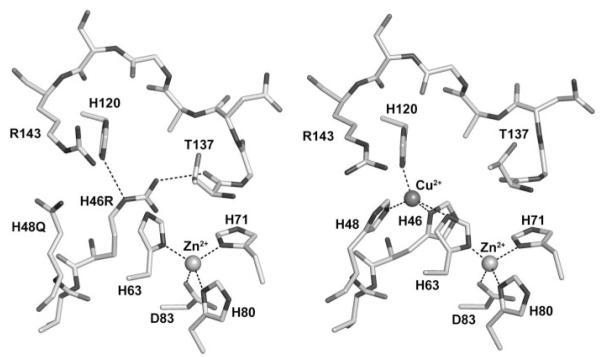
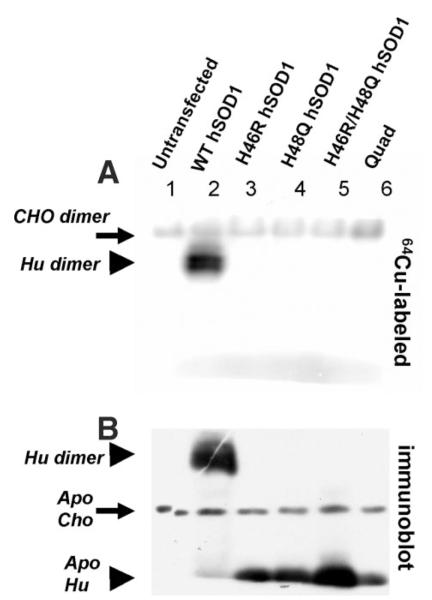
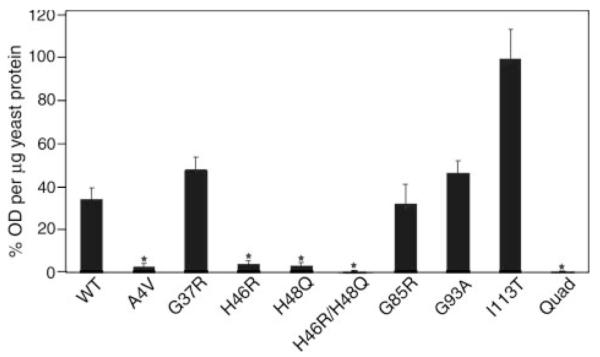
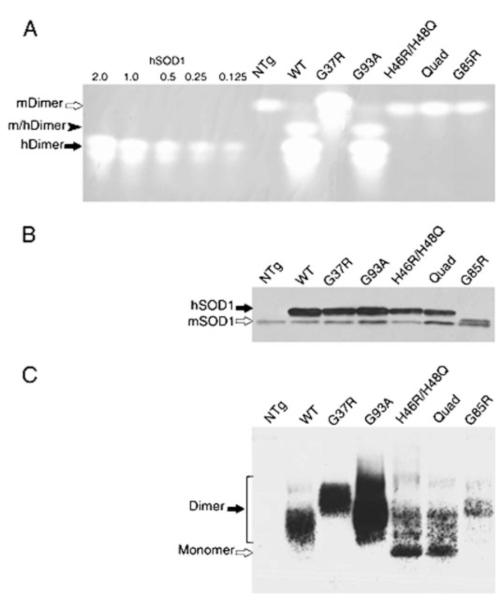
A subset of superoxide dismutase 1 (Cu/Zn-SOD1) mutants that cause familial amyotrophic lateral sclerosis (FALS) have heightened reactivity with (-)ONOO and H(2)O(2) in vitro. This reactivity requires a copper ion bound in the active site and is a suggested mechanism of motor neuron injury. However, we have found that transgenic mice that express SOD1-H46R/H48Q, which combines natural FALS mutations at ligands for copper and which is inactive, develop motor neuron disease. Using a direct radioactive copper incorporation assay in transfected cells and the established tools of single crystal x-ray diffraction, we now demonstrate that this variant does not stably bind copper. We find that single mutations at copper ligands, including H46R, H48Q, and a quadruple mutant H46R/H48Q/H63G/H120G, also diminish the binding of radioactive copper. Further, using native polyacrylamide gel electrophoresis and a yeast two-hybrid assay, the binding of copper was found to be related to the formation of the stable dimeric enzyme. Collectively, our data demonstrate a relationship between copper and assembly of SOD1 into stable dimers and also define disease-causing SOD1 mutants that are unlikely to robustly produce toxic radicals via copper-mediated chemistry.
Figures
References
-
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr. Nature. 1993;362:59–62. - PubMed
-
- Andersen PM, Morita M, Brown RH., Jr. In: Amyotrophic Lateral Sclerosis. Brown RH Jr., Meininger V, Swash M, editors. Martin Dunitz Ltd.; London: 2000. pp. 223–250.
-
- Cleveland DW, Rothstein JD. Nat. Rev. Neurosci. 2001;2:806–819. - PubMed
-
- Hart PJ. Curr. Opin. Chem. Biol. 2006;10:131–138. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
