

Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia
- PMID: 17093095
- PMCID: PMC6674790
- DOI: 10.1523/JNEUROSCI.3581-06.2006
Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia
Abstract
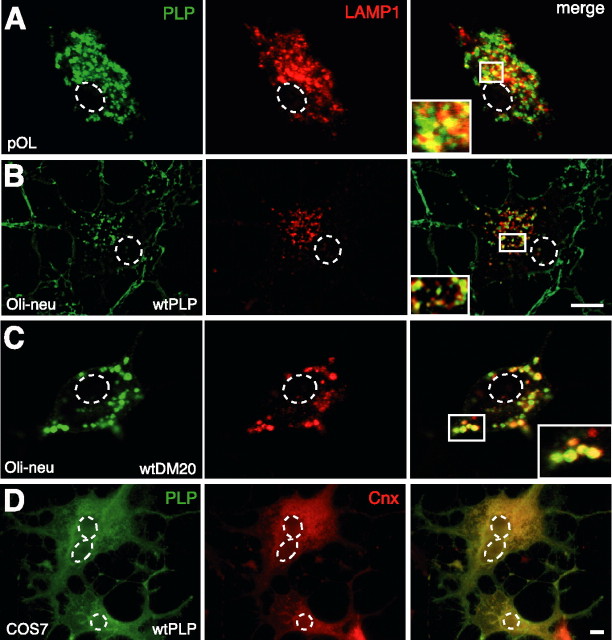
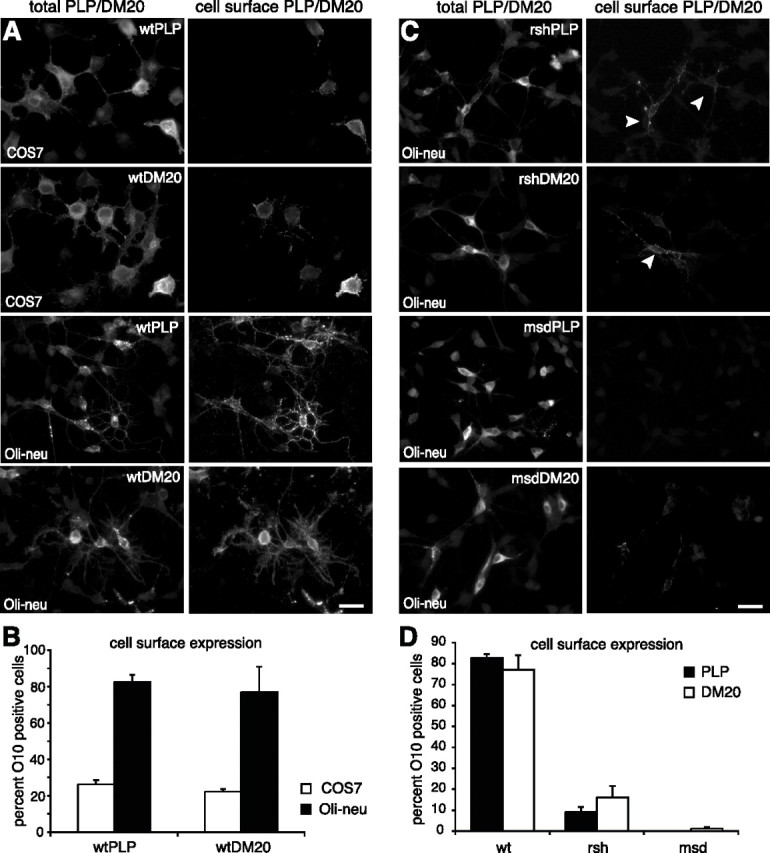
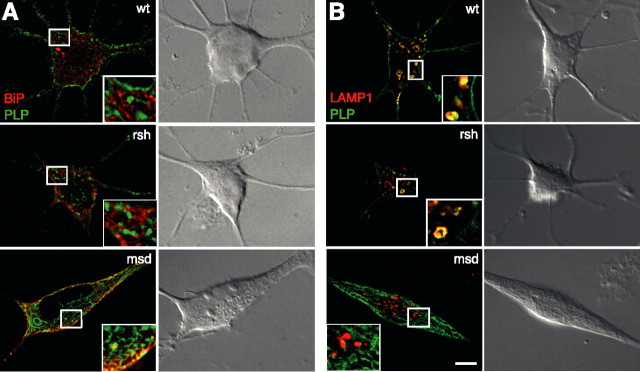
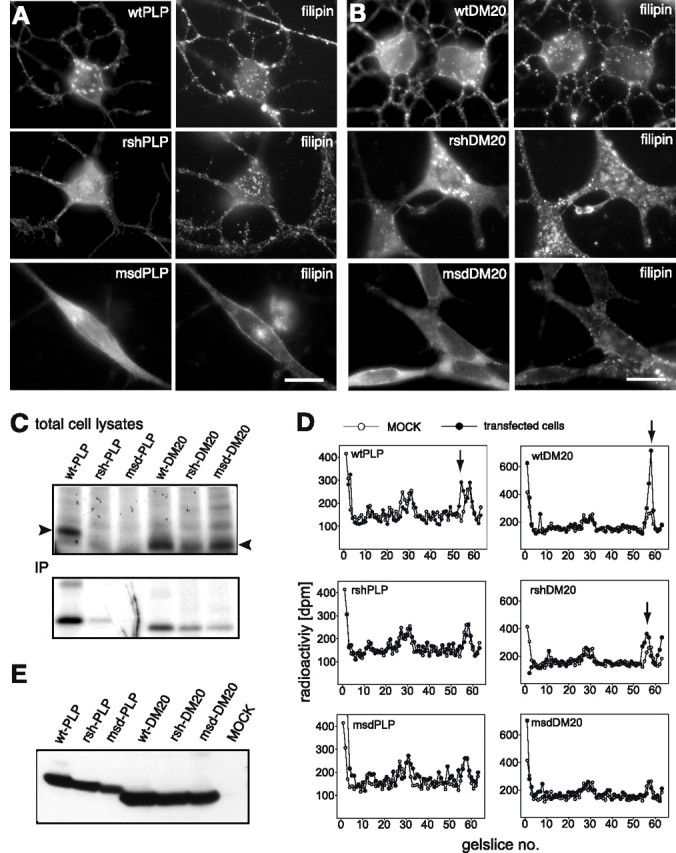
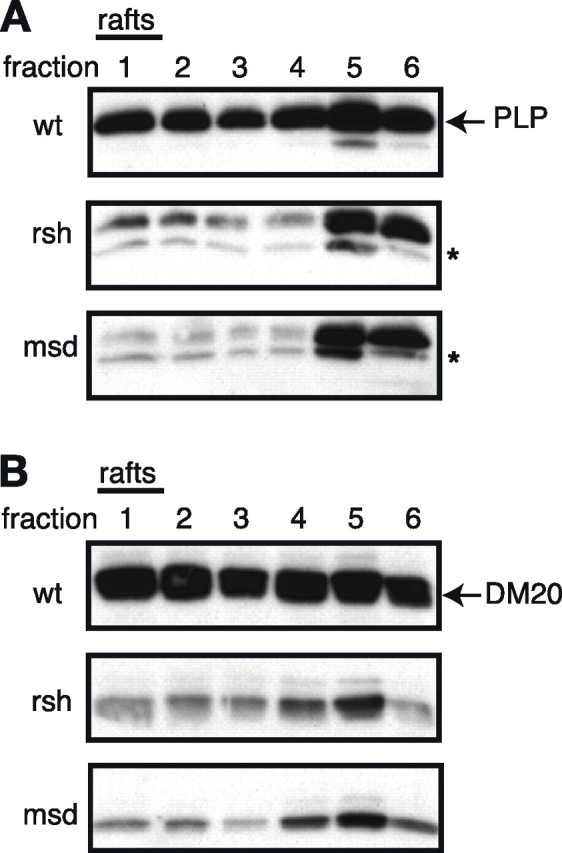
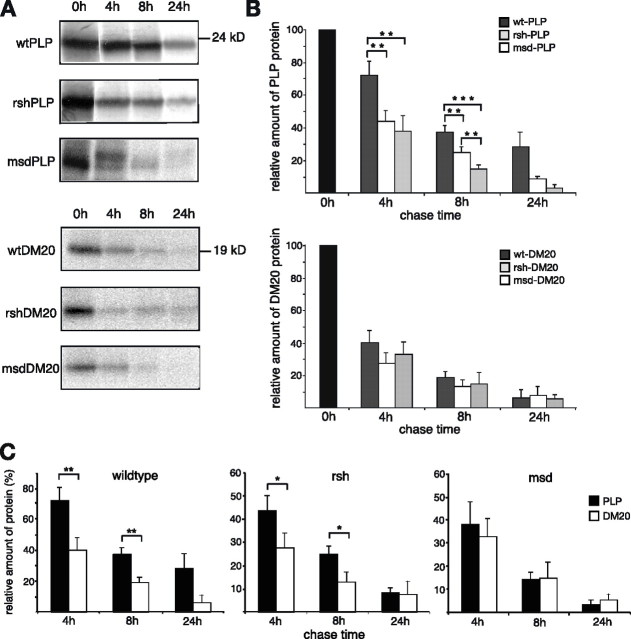
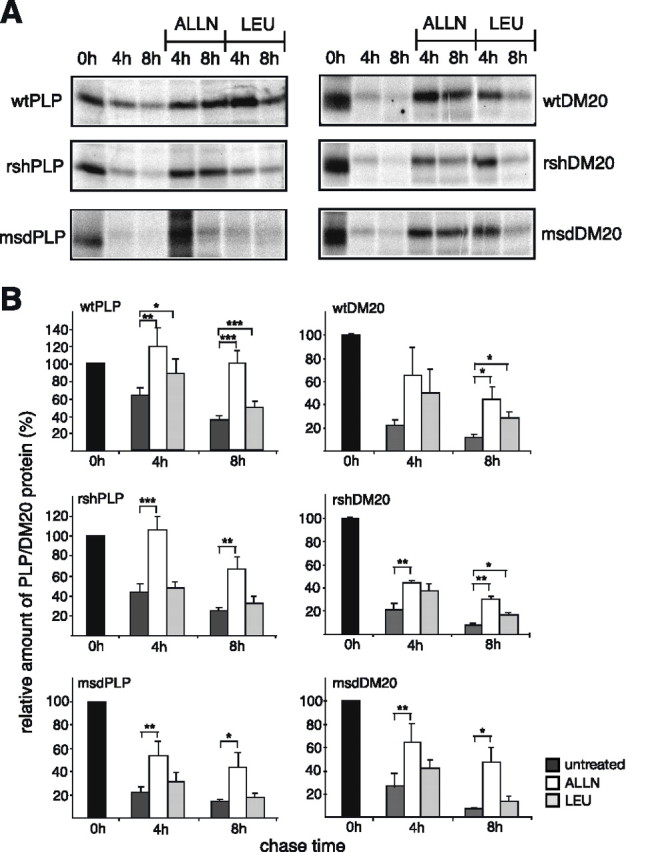
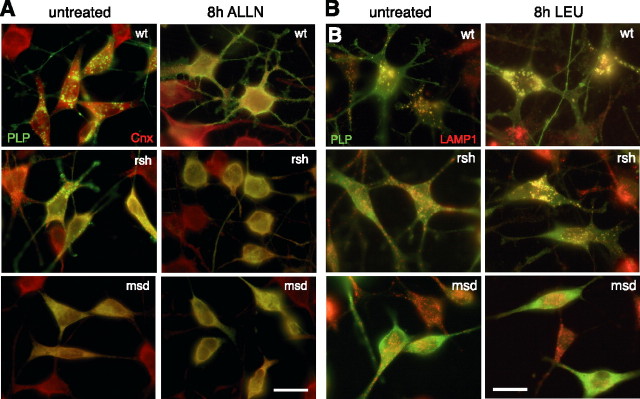
Missense mutations in the human PLP1 gene lead to dysmyelinating diseases with a broad range of clinical severity, ranging from severe Pelizaeus-Merzbacher disease (PMD) to milder spastic paraplegia type 2 (SPG-2). The molecular pathology has been generally attributed to endoplasmic reticulum (ER) retention of misfolded proteolipid protein (PLP) (and its splice isoform DM20) and induction of the unfolded protein response. As opposed to previous studies of heterologous expression systems, we have analyzed PLP/DM20 trafficking in oligodendroglial cells, thereby revealing differences between PMD and SPG-2-associated PLP/DM20 isoforms. PLP(A242V) and DM20(A242V) (jimpy-msd in mice), associated with severe PMD-like phenotype in vivo, were not only retained in the ER but also interfered with oligodendroglial process formation. In contrast, glial cells expressing SPG-2-associated PLP(I186T) or DM20(I186T) (rumpshaker in mice) developed processes, and mutant PLP/DM20 reached a late endosomal/lysosomal compartment. Unexpectedly, PLP/DM20 with either substitution exhibited impaired cholesterol binding, and the association with lipid raft microdomains was strongly reduced. Turnover analysis demonstrated that mutant PLP was rapidly degraded in oligodendroglial cells, with half-lives for PLP > PLP(I186T) > PLP(A242V). Protein degradation was specifically sensitive to proteasome inhibition, although PLP/DM20(I186T) degradation was also affected by inhibition of lysosomal enzymes. We conclude that, in addition to ER retention and unfolded protein response (UPR) induction, impaired cholesterol binding and lipid raft association are characteristic cellular defects of PLP1-missense mutations. Mutant protein is rapidly cleared and does not accumulate in oligodendroglial cells. Whereas UPR-induced cell death governs the PMD phenotype of the msd mutation, we propose that impaired cholesterol and lipid raft interaction of the rsh protein may contribute to the dysmyelination observed in SPG-2.
Figures
Similar articles
-
Phenotypic severity of murine Plp mutants reflects in vivo and in vitro variations in transport of PLP isoproteins.Glia. 1997 Aug;20(4):322-32. doi: 10.1002/(sici)1098-1136(199708)20:4<322::aid-glia5>3.0.co;2-7. Glia. 1997. PMID: 9262236
-
PLP overexpression perturbs myelin protein composition and myelination in a mouse model of Pelizaeus-Merzbacher disease.Glia. 2007 Mar;55(4):341-51. doi: 10.1002/glia.20465. Glia. 2007. PMID: 17133418
-
Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease.J Cell Biol. 2002 Apr 15;157(2):327-36. doi: 10.1083/jcb.200110138. Epub 2002 Apr 15. J Cell Biol. 2002. PMID: 11956232 Free PMC article.
-
Current concepts of PLP and its role in the nervous system.Microsc Res Tech. 1998 Jun 1;41(5):344-58. doi: 10.1002/(SICI)1097-0029(19980601)41:5<344::AID-JEMT2>3.0.CO;2-Q. Microsc Res Tech. 1998. PMID: 9672418 Review.
-
Pelizaeus-Merzbacher disease.J Neuropathol Exp Neurol. 2002 Sep;61(9):747-59. doi: 10.1093/jnen/61.9.747. J Neuropathol Exp Neurol. 2002. PMID: 12230321 Review.
Cited by
-
Cholesterol Biosynthesis Supports Myelin Gene Expression and Axon Ensheathment through Modulation of P13K/Akt/mTor Signaling.J Neurosci. 2016 Jul 20;36(29):7628-39. doi: 10.1523/JNEUROSCI.0726-16.2016. J Neurosci. 2016. PMID: 27445141 Free PMC article.
-
Cell-specific loss of kappa-opioid receptors in oligodendrocytes of the dysmyelinating jimpy mouse.Neurosci Lett. 2009 Feb 20;451(2):114-8. doi: 10.1016/j.neulet.2008.12.022. Epub 2008 Dec 24. Neurosci Lett. 2009. PMID: 19110031 Free PMC article.
-
Differences in endoplasmic-reticulum quality control determine the cellular response to disease-associated mutants of proteolipid protein.J Cell Sci. 2009 Nov 1;122(Pt 21):3942-53. doi: 10.1242/jcs.055160. Epub 2009 Oct 13. J Cell Sci. 2009. PMID: 19825935 Free PMC article.
-
Chemical Screening Identifies Enhancers of Mutant Oligodendrocyte Survival and Unmasks a Distinct Pathological Phase in Pelizaeus-Merzbacher Disease.Stem Cell Reports. 2018 Sep 11;11(3):711-726. doi: 10.1016/j.stemcr.2018.07.015. Epub 2018 Aug 23. Stem Cell Reports. 2018. PMID: 30146490 Free PMC article.
-
A single gene defect causing claustrophobia.Transl Psychiatry. 2013 Apr 30;3(4):e254. doi: 10.1038/tp.2013.28. Transl Psychiatry. 2013. PMID: 23632458 Free PMC article.
References
-
- Al-Saktawi K, McLaughlin M, Klugmann M, Schneider A, Barrie JA, McCulloch MC, Montague P, Kirkham D, Nave KA, Griffiths IR. Genetic background determines phenotypic severity of the PLP-rumpshaker mutation. J Neurosci Res. 2003;72:12–24. - PubMed
-
- Duncan ID. The PLP mutants from mouse to man. J Neurol Sci. 2005;228:204–205. - PubMed
-
- Forman MS, Lee VY, Trojanowski JQ. “Unfolding” pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–410. - PubMed
-
- Garbern JY. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J Neurol Sci. 2005;228:201–203. - PubMed
-
- Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, van der Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffiths IR. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551–561. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials