

Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts
- PMID: 17098208
- PMCID: PMC1847786
- DOI: 10.1016/j.abb.2006.09.010
Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts
Abstract
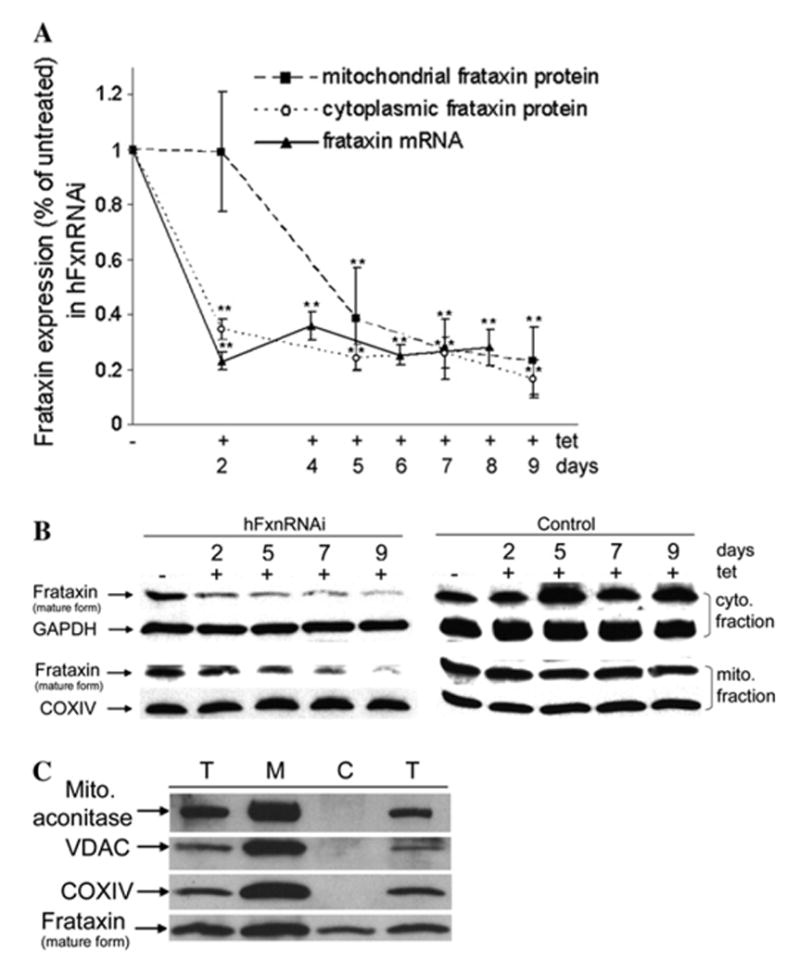
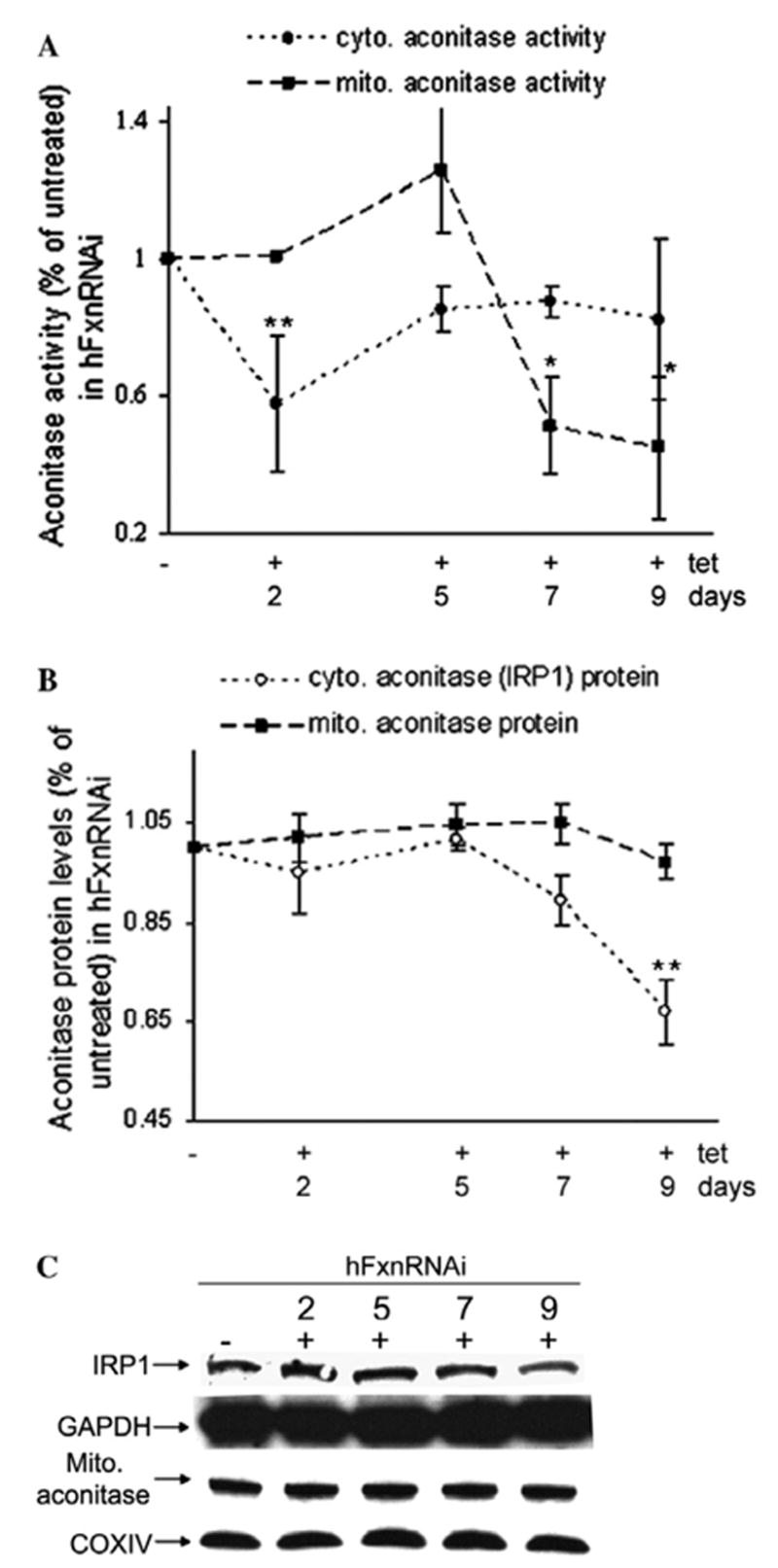
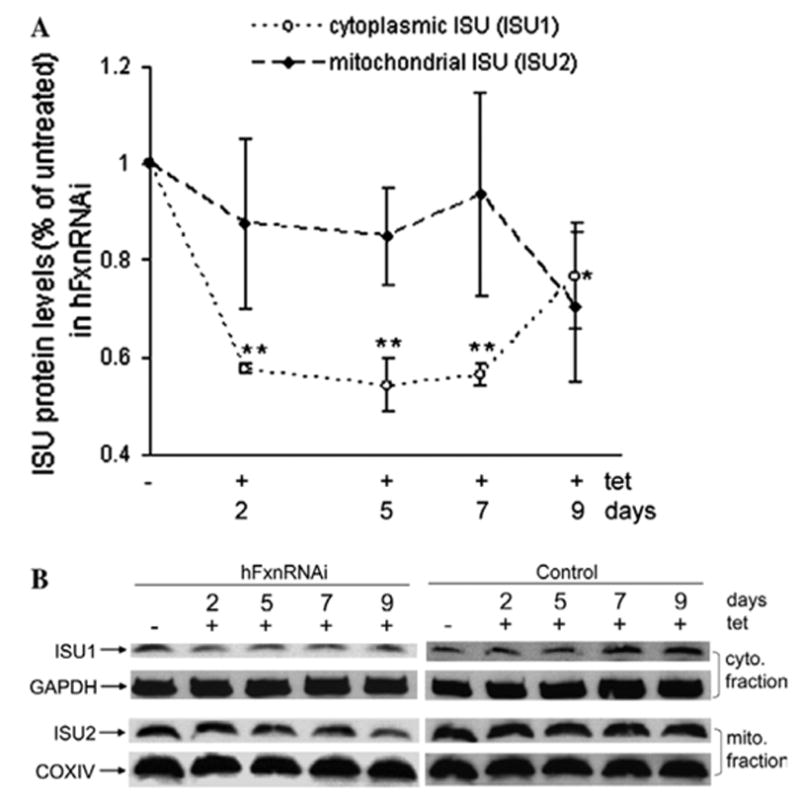
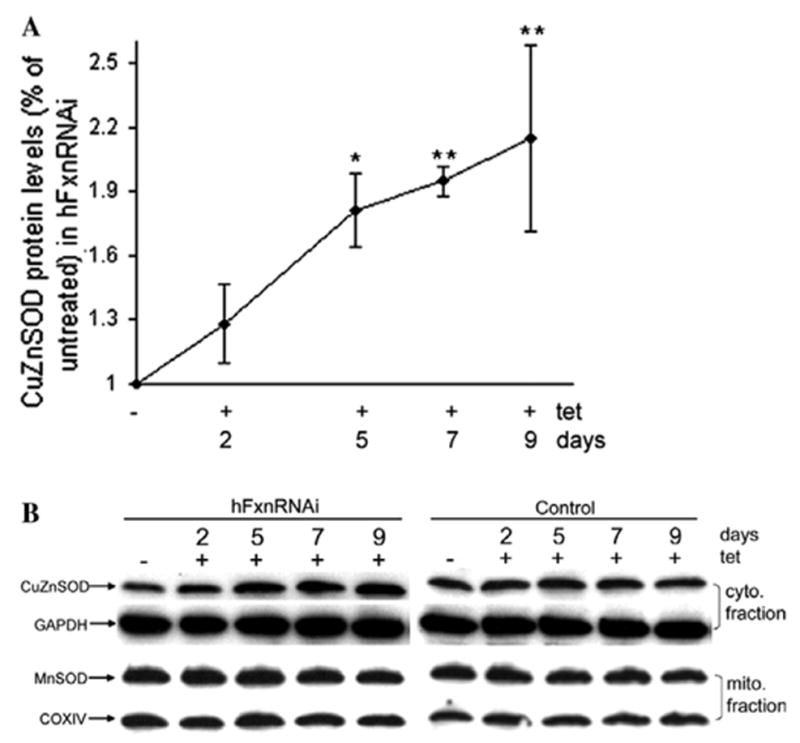
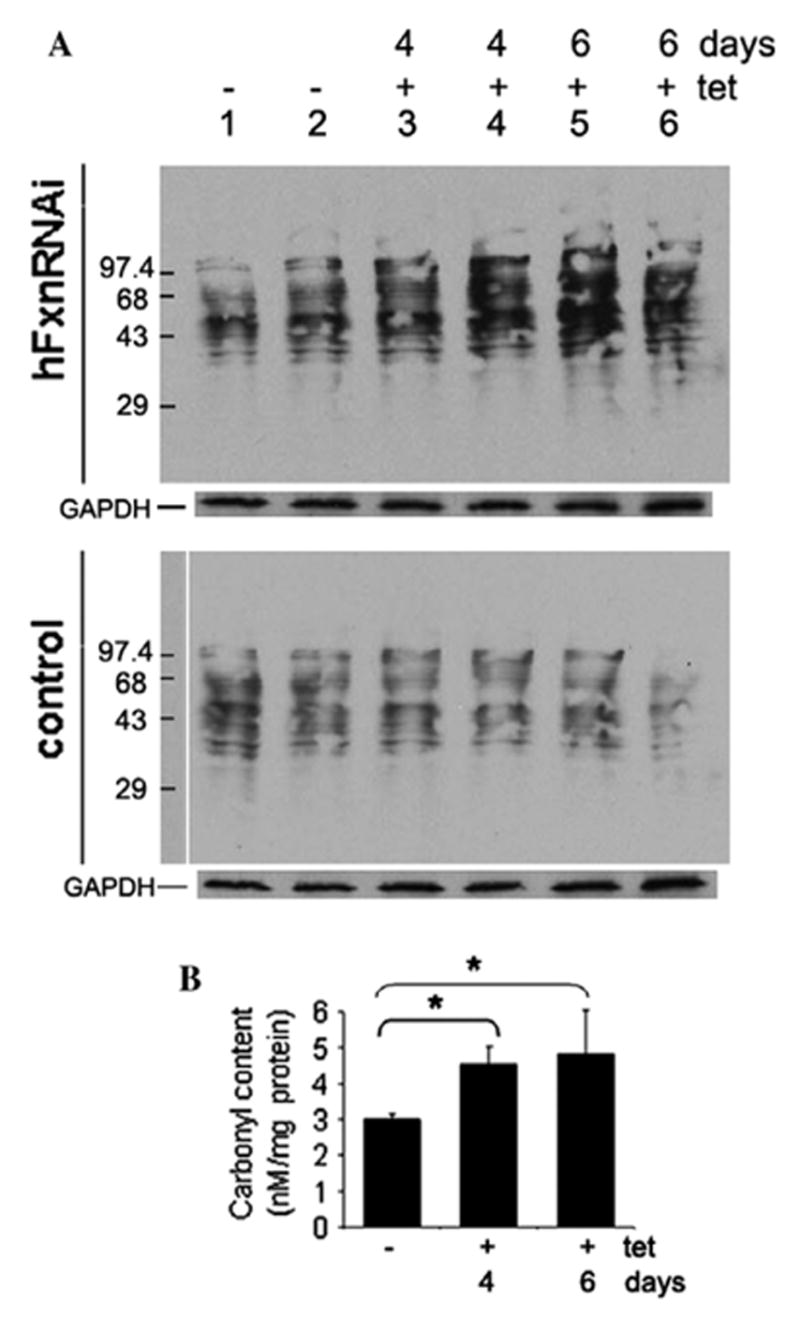
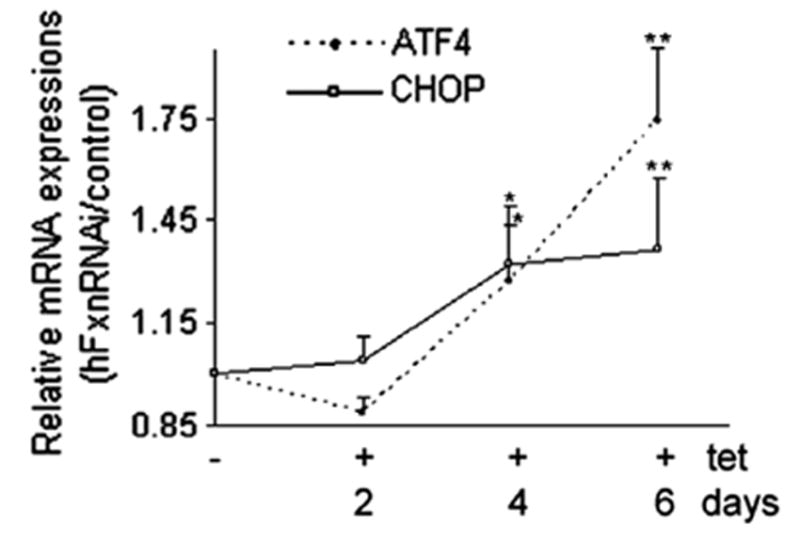
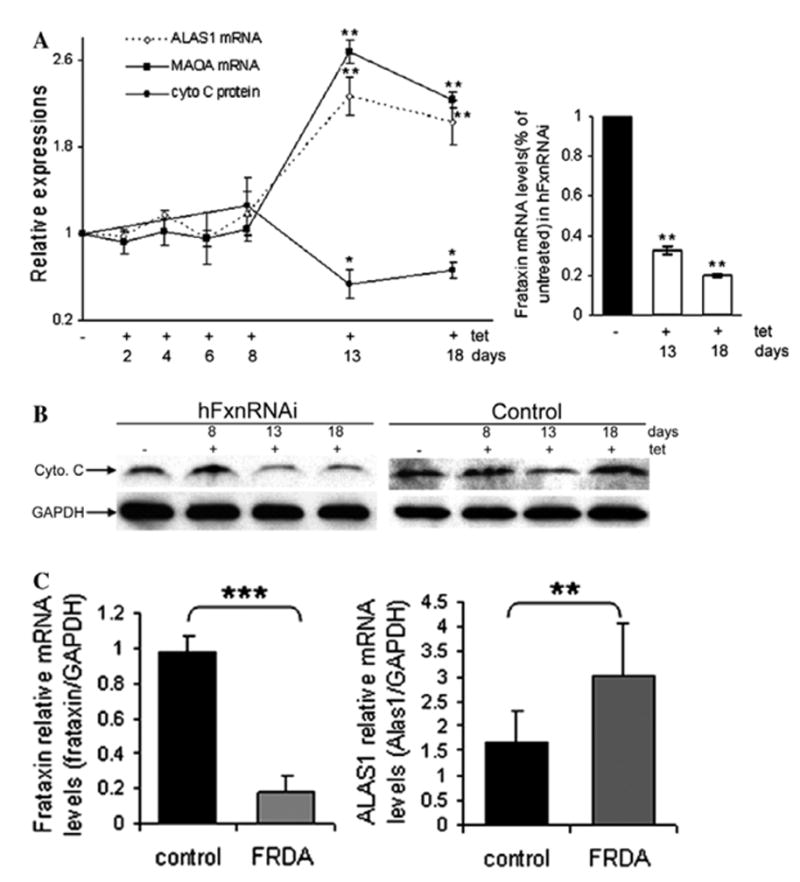
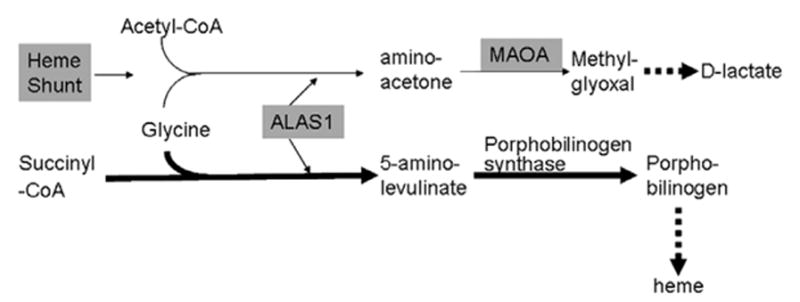
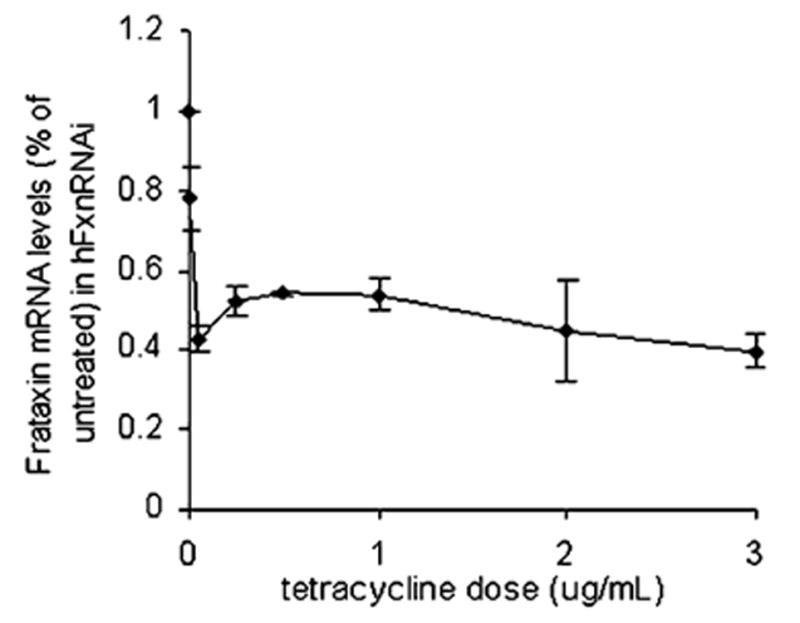
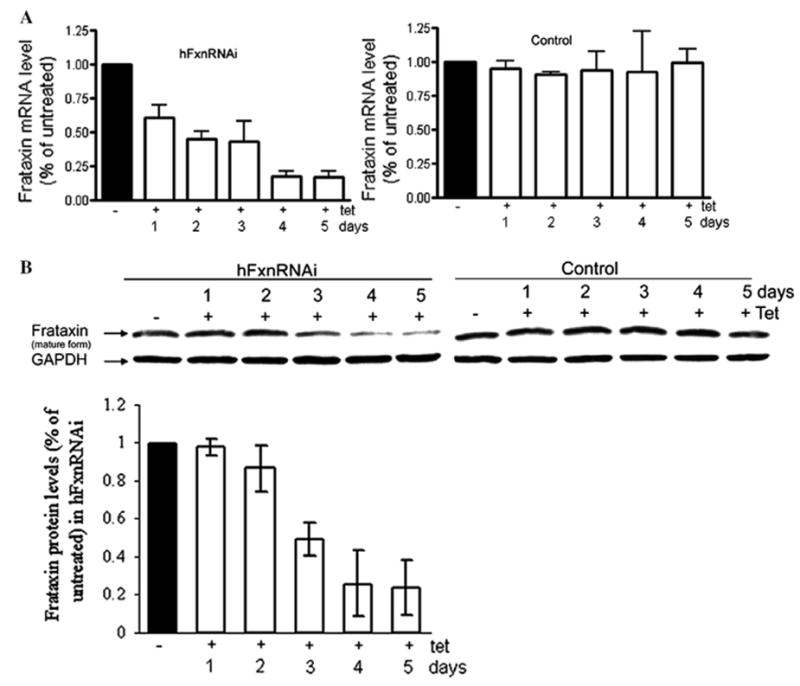
Frataxin protein deficiency causes the neurodegenerative disease Friedreich ataxia. We used inducible siRNA to order the consequences of frataxin deficiency that we and others have previously observed. The earliest consequence of frataxin deficiency was a defect in cytoplasmic iron-sulfur proteins. In the second phase, protein oxidative damage increased, and CuZnSOD was induced, as was the unfolded protein response (UPR), long before any decline in mitochondrial aconitase activity. In the third phase, mitochondrial aconitase activity declined. And in the fourth phase, coincident with the decrease in heme-containing cytochrome c protein, a transcriptional induction of the heme-dependent transcripts ALAS1 and MAOA occurred. These observations suggest that the earliest consequences of frataxin deficiency occur in ISC proteins of the cytoplasm, resulting in oxidative damage and stress and activation of the unfolded protein response which has been associated with neurological disease, and that later consequences involve mitochondrial iron-sulfur cluster deficiency, heme deficiency, and then increased heme biosynthesis.
Figures
References
-
- Harding AE. The Hereditary Ataxias and Related Disorders. Churchill Livingstone: Edinburgh; 1984.
-
- Harding AE. The inherited ataxias. Adv Neurol. 1988;48:37–46. - PubMed
-
- Johnson WG. Friedreich ataxia. Clin Neurosci. 1995;3:33–38. - PubMed
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
