

The role of TGF-beta signaling in myocardial infarction and cardiac remodeling
- PMID: 17109837
- PMCID: PMC1924687
- DOI: 10.1016/j.cardiores.2006.10.002
The role of TGF-beta signaling in myocardial infarction and cardiac remodeling
Abstract
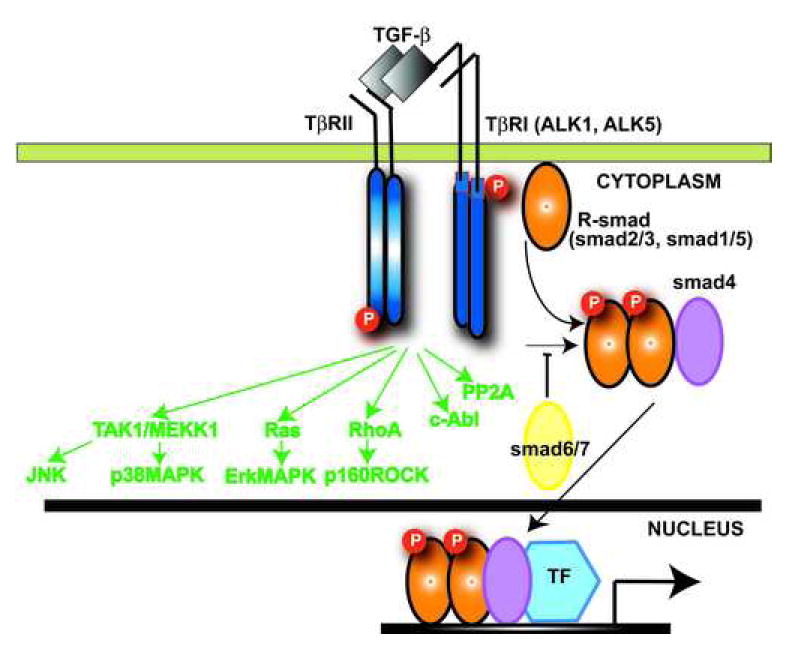
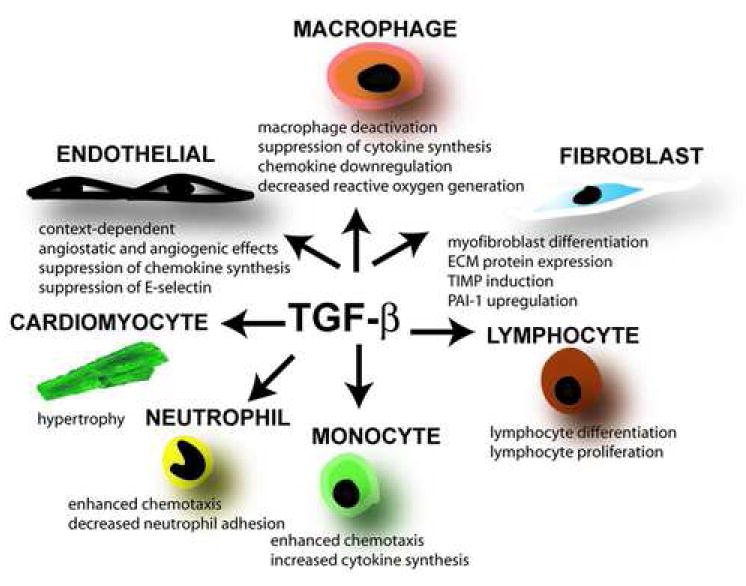
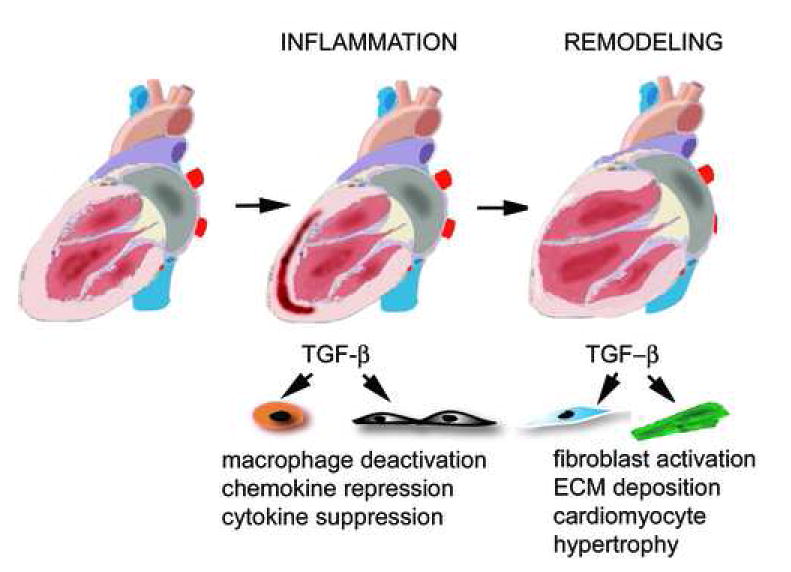
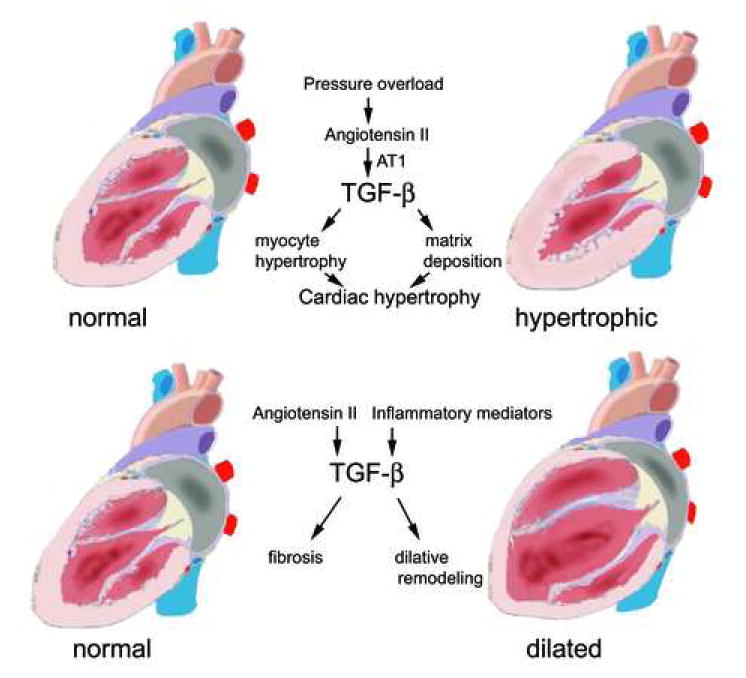
Transforming Growth Factor (TGF)-beta is markedly induced and rapidly activated in the infarcted myocardium. However, understanding of the exact role of TGF-beta signaling in the infarcted and remodeling heart has been hampered by the complex and unusual biology of TGF-beta activation and by the diversity of its effects eliciting multiple, and often opposing cellular responses. Experimental studies suggest that TGF-beta signaling may be crucial for repression of inflammatory gene synthesis in healing infarcts mediating resolution of the inflammatory infiltrate. In addition, TGF-beta may play an important role in modulating fibroblast phenotype and gene expression, promoting extracellular matrix deposition in the infarct by upregulating collagen and fibronectin synthesis and by decreasing matrix degradation through induction of protease inhibitors. TGF-beta is also a key mediator in the pathogenesis of hypertrophic and dilative ventricular remodeling by stimulating cardiomyocyte growth and by inducing interstitial fibrosis. In this review we summarize the current knowledge on the role of TGF-beta in infarct healing and cardiac remodeling.
Figures
References
-
- Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. - PubMed
-
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–61. - PubMed
-
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–24. - PubMed
-
- Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–45. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
