

Generalization of the Gaussian electrostatic model: extension to arbitrary angular momentum, distributed multipoles, and speedup with reciprocal space methods
- PMID: 17115732
- PMCID: PMC2080839
- DOI: 10.1063/1.2363374
Generalization of the Gaussian electrostatic model: extension to arbitrary angular momentum, distributed multipoles, and speedup with reciprocal space methods
Abstract
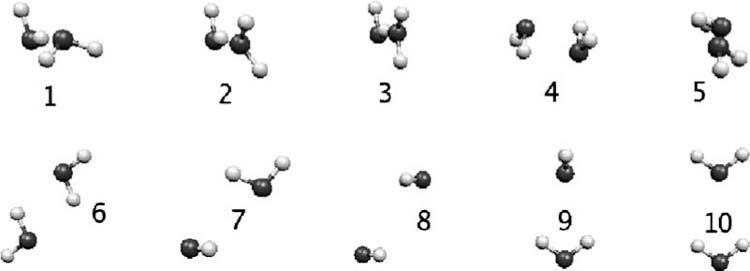
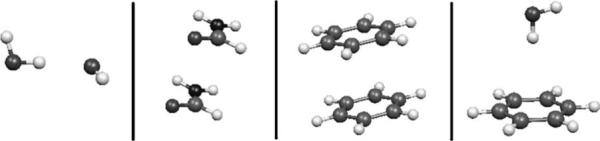
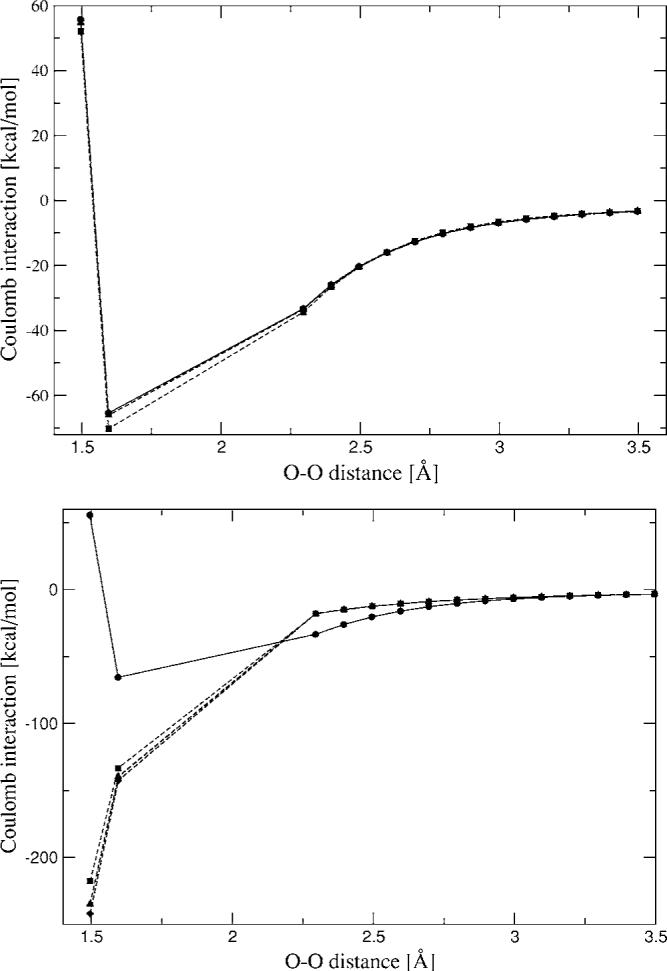
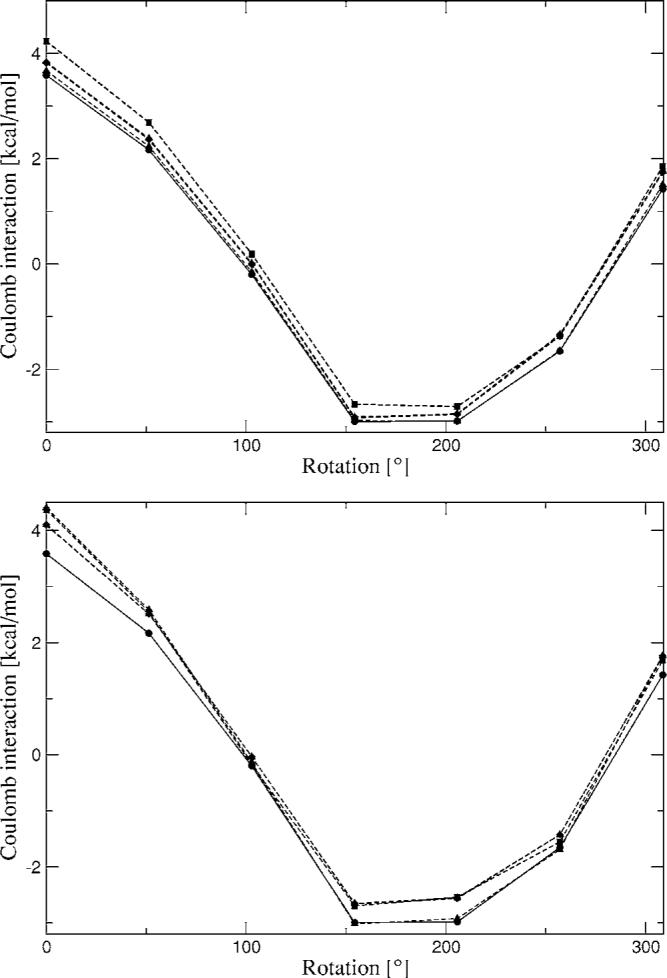
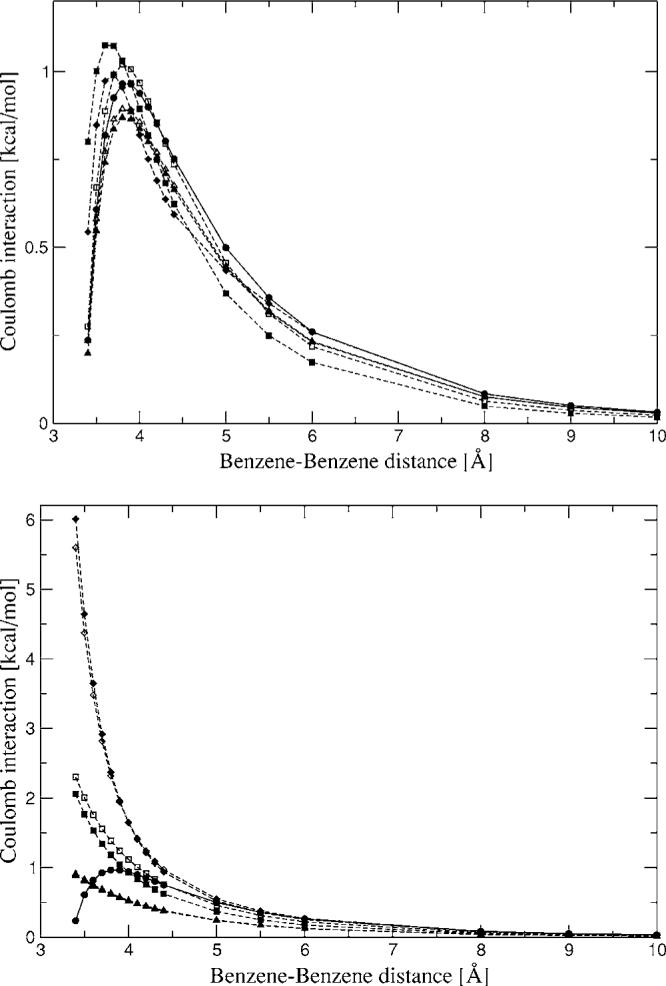
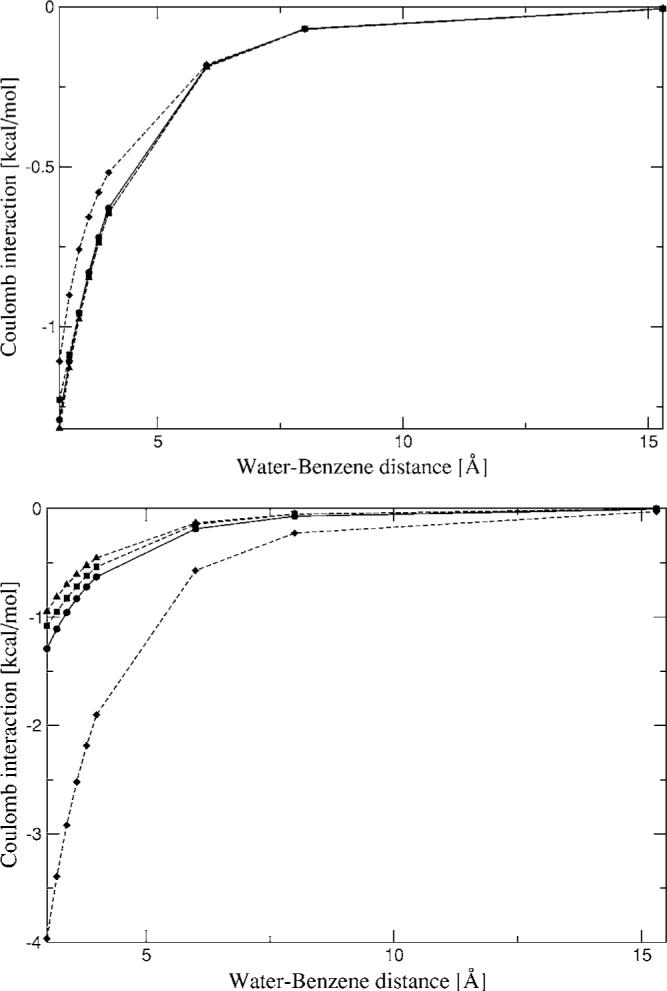
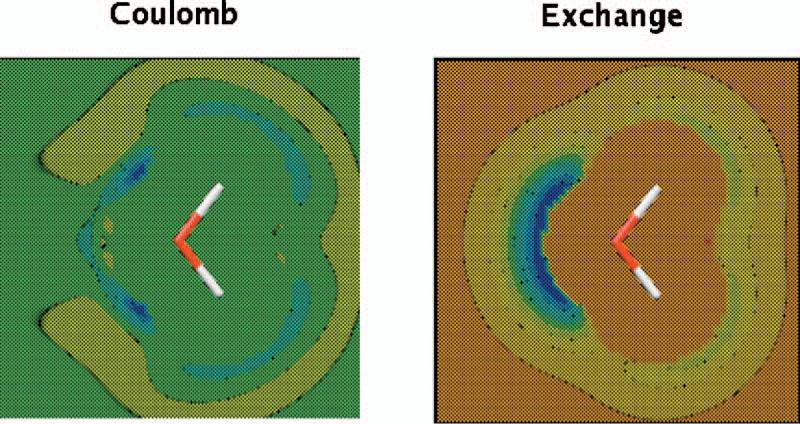
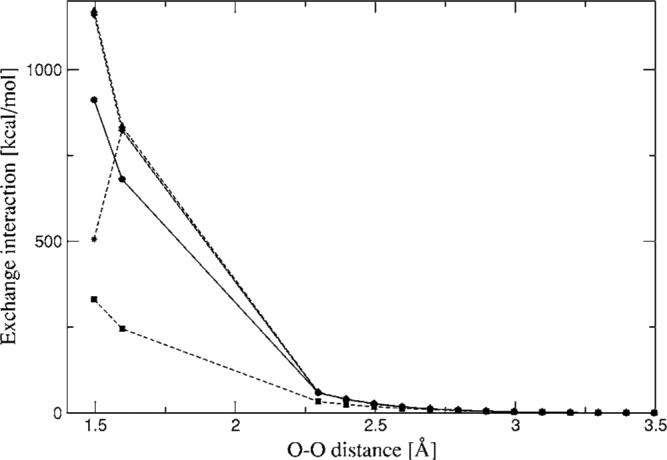
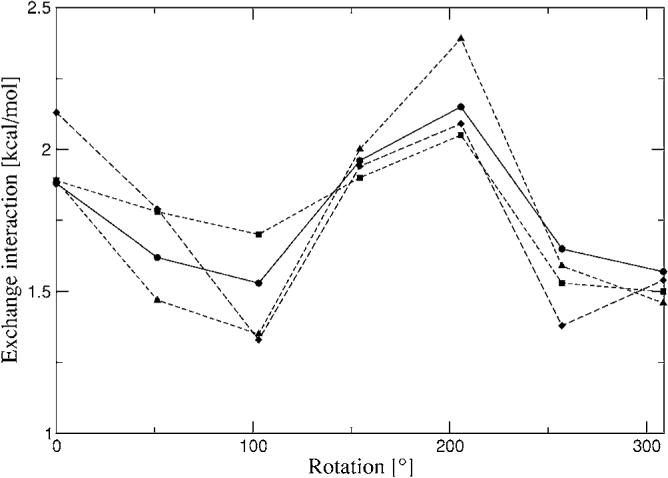
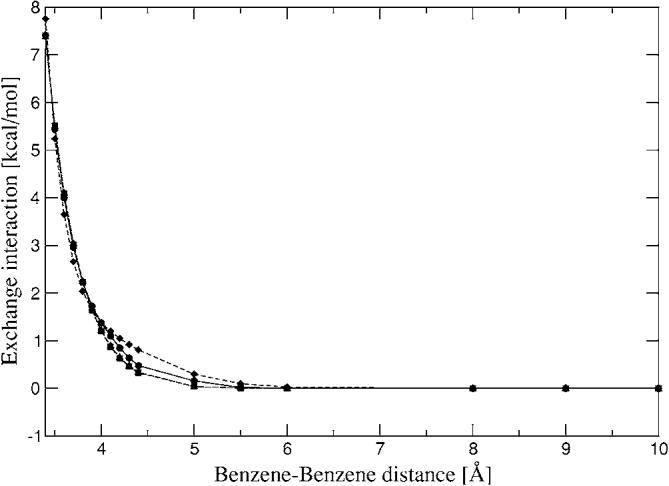
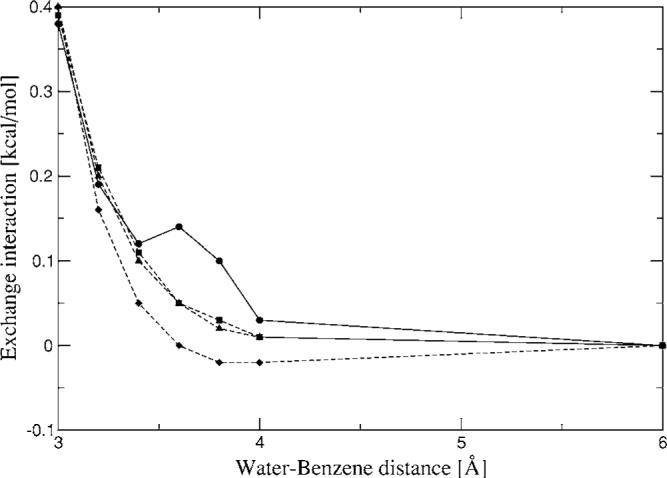
The simulation of biological systems by means of current empirical force fields presents shortcomings due to their lack of accuracy, especially in the description of the nonbonded terms. We have previously introduced a force field based on density fitting termed the Gaussian electrostatic model-0 (GEM-0) J.-P. Piquemal et al. [J. Chem. Phys. 124, 104101 (2006)] that improves the description of the nonbonded interactions. GEM-0 relies on density fitting methodology to reproduce each contribution of the constrained space orbital variation (CSOV) energy decomposition scheme, by expanding the electronic density of the molecule in s-type Gaussian functions centered at specific sites. In the present contribution we extend the Coulomb and exchange components of the force field to auxiliary basis sets of arbitrary angular momentum. Since the basis functions with higher angular momentum have directionality, a reference molecular frame (local frame) formalism is employed for the rotation of the fitted expansion coefficients. In all cases the intermolecular interaction energies are calculated by means of Hermite Gaussian functions using the McMurchie-Davidson [J. Comput. Phys. 26, 218 (1978)] recursion to calculate all the required integrals. Furthermore, the use of Hermite Gaussian functions allows a point multipole decomposition determination at each expansion site. Additionally, the issue of computational speed is investigated by reciprocal space based formalisms which include the particle mesh Ewald (PME) and fast Fourier-Poisson (FFP) methods. Frozen-core (Coulomb and exchange-repulsion) intermolecular interaction results for ten stationary points on the water dimer potential-energy surface, as well as a one-dimensional surface scan for the canonical water dimer, formamide, stacked benzene, and benzene water dimers, are presented. All results show reasonable agreement with the corresponding CSOV calculated reference contributions, around 0.1 and 0.15 kcal/mol error for Coulomb and exchange, respectively. Timing results for single Coulomb energy-force calculations for (H(2)O)(n), n=64, 128, 256, 512, and 1024, in periodic boundary conditions with PME and FFP at two different rms force tolerances are also presented. For the small and intermediate auxiliaries, PME shows faster times than FFP at both accuracies and the advantage of PME widens at higher accuracy, while for the largest auxiliary, the opposite occurs.
Figures
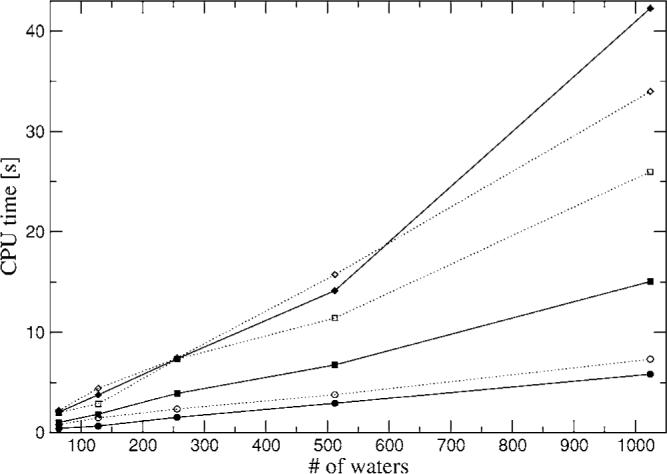
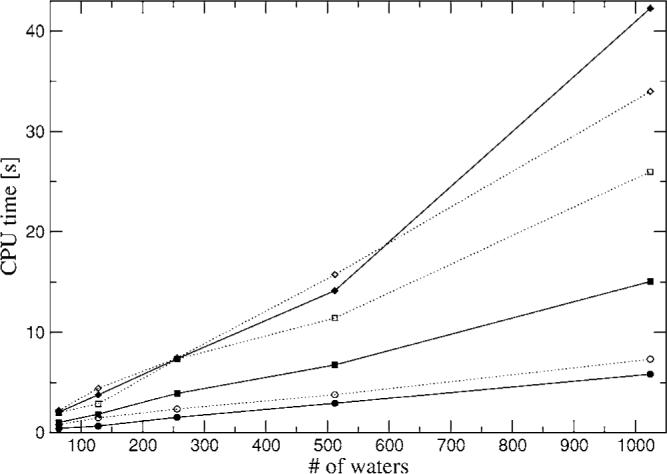
References
-
- Jorgensen WL, Tirado-Rives J. J. Comput. Chem. 2005;26:1669. - PubMed
-
- van der Spoel D, Lindahl E, Hess B, Groenhoff G, Mark AE, Berensen HJC. J. Comput. Chem. 2005;26:1701. - PubMed
-
- Christen M, Hünenberger PH, Bakowies D, et al. J. Comput. Chem. 2005;26:1719. - PubMed
-
- MacKerrell AD, Jr., Brooks B, Brooks CL, III, Roux NB, Won Y, Karplus M. Encyclopedia of Computational Chemistry. Wiley; NewYork, NY: 1998.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials