

Cardiac dysfunction in the R6/2 mouse model of Huntington's disease
- PMID: 17126554
- PMCID: PMC1850107
- DOI: 10.1016/j.nbd.2006.09.016
Cardiac dysfunction in the R6/2 mouse model of Huntington's disease
Abstract
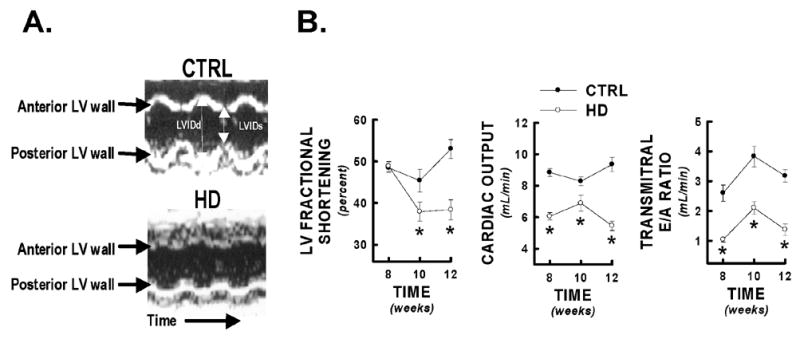
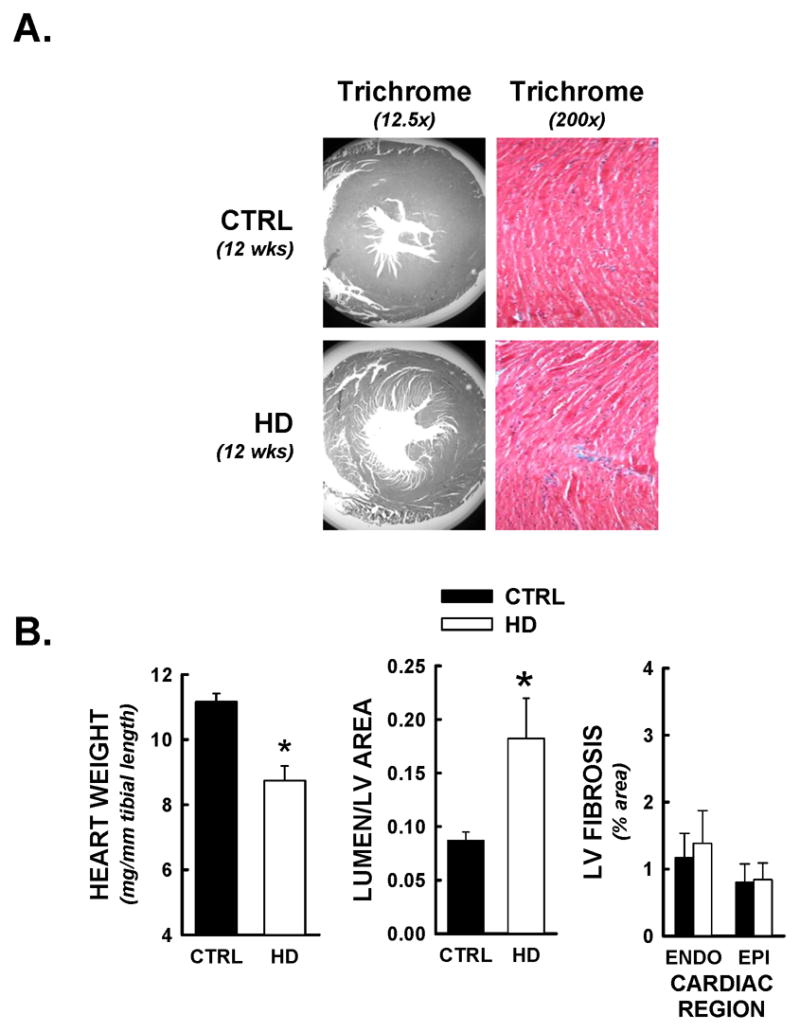
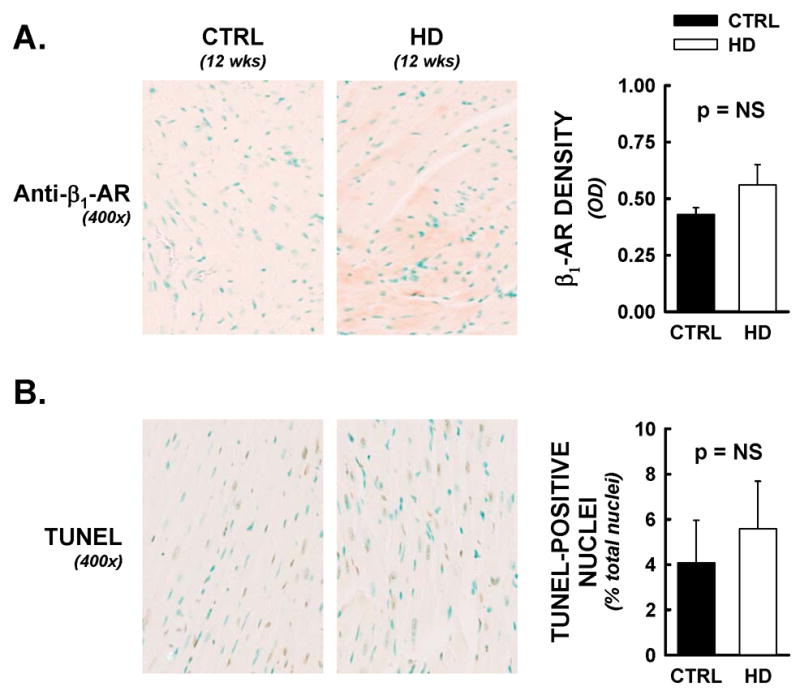
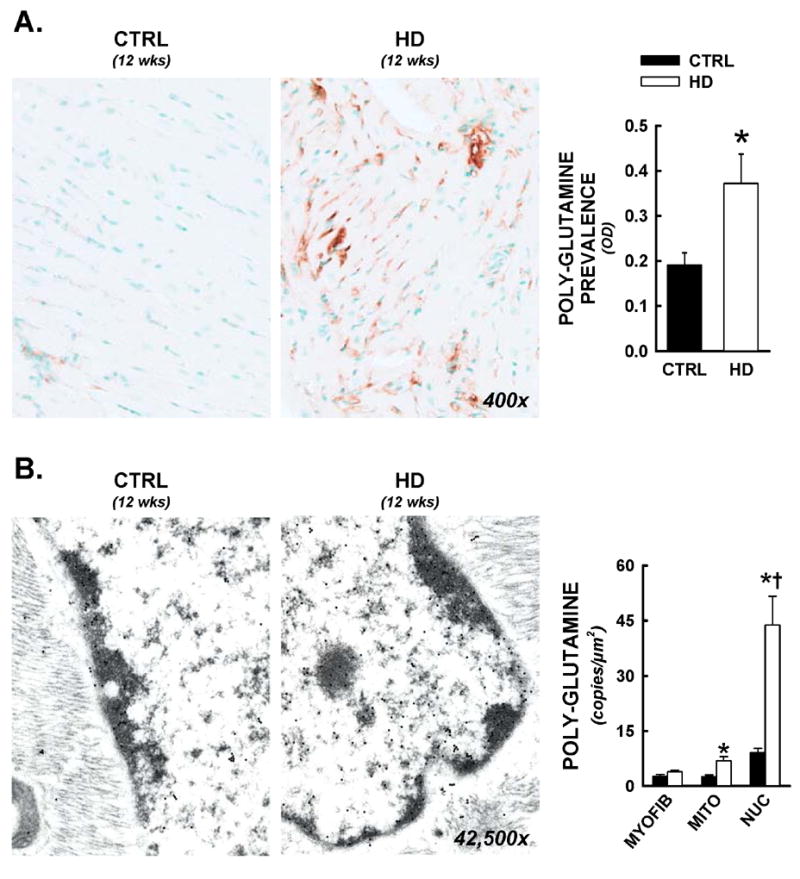
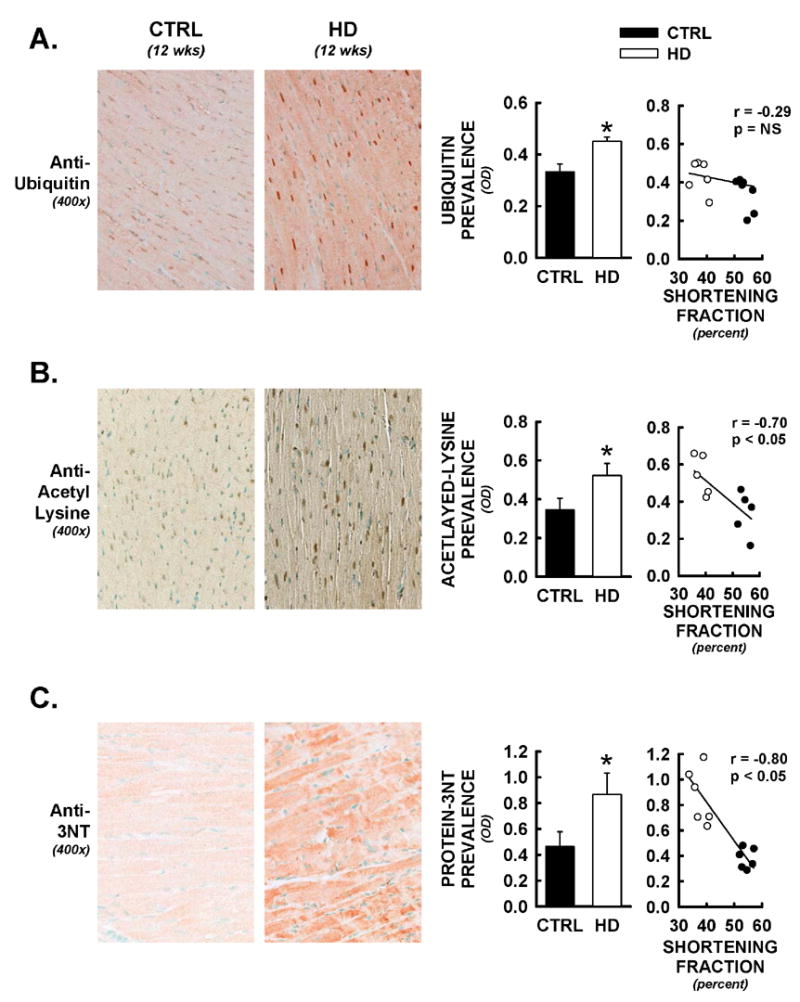
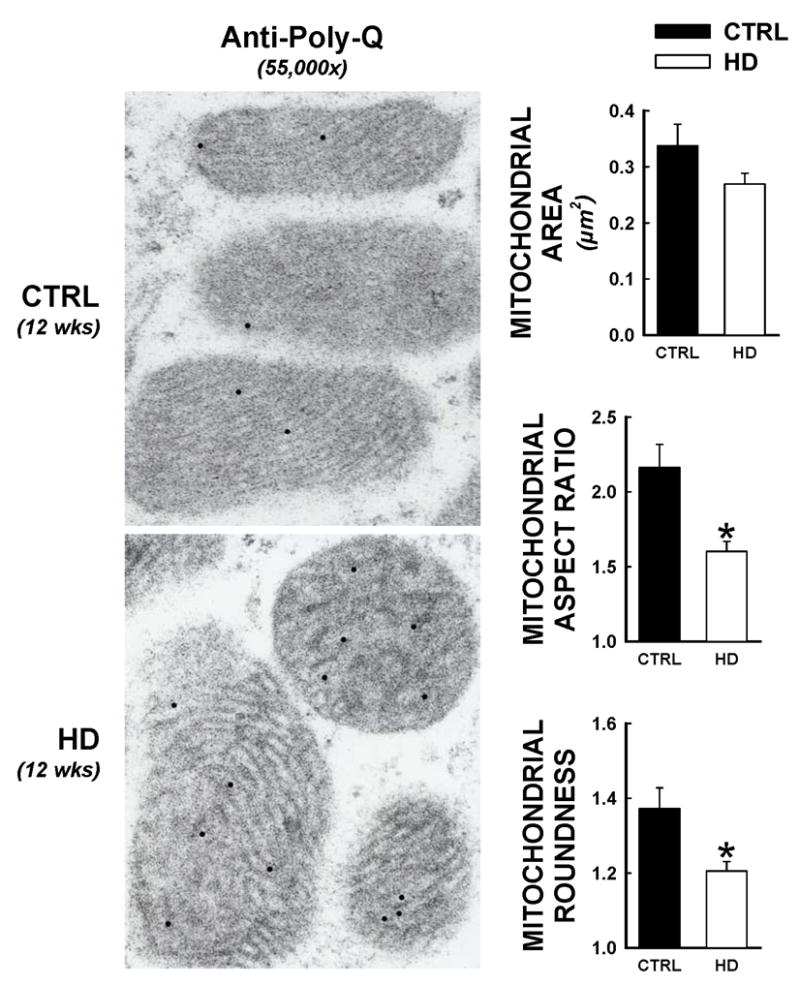
Recent evidence suggests that mutant huntingtin protein-induced energetic perturbations contribute to neuronal dysfunction in Huntington's disease (HD). Given the ubiquitous expression of huntingtin, other cell types with high energetic burden may be at risk for HD-related dysfunction. Early-onset cardiovascular disease is the second leading cause of death in HD patients; a direct role for mutant huntingtin in this phenomenon remains unevaluated. Here we tested the hypothesis that expression of mutant huntingtin is sufficient to induce cardiac dysfunction, using a well-described transgenic model of HD (line R6/2). R6/2 mice developed cardiac dysfunction by 8 weeks of age, progressing to severe failure at 12 weeks, assessed by echocardiography. Limited evidence of cardiac remodeling (e.g. hypertrophy, fibrosis, apoptosis, beta(1) adrenergic receptor downregulation) was observed. Immunogold electron microscopy demonstrated significant elevations in nuclear and mitochondrial polyglutamine presence in the R6/2 myocyte. Significant alterations in mitochondrial ultrastructure were seen, consistent with metabolic stress. Increased cardiac lysine acetylation and protein nitration were observed and were each significantly associated with impairments in cardiac performance. These data demonstrate that mutant huntingtin expression has potent cardiotoxic effects; cardiac failure may be a significant complication of this important experimental model of HD. Investigation of the potential cardiotropic effects of mutant huntingtin in humans may be warranted.
Figures
References
-
- Andreassen OA, Ferrante RJ, Dedeoglu A, Beal MF. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport. 2001a;12:3371–3373. - PubMed
-
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol Dis. 2001b;8:479–491. - PubMed
-
- Arenas J, Campos Y, Ribacoba R, Martin MA, Rubio JC, Ablanedo P, Cabello A. Complex I defect in muscle from patients with Huntington’s disease. Annals of Neurology. 1998;43:397–400. - PubMed
-
- Bates G, Harper P, Jones L, editors. Huntington’s Disease. 3. Oxford ; New York: Oxford University Press; 2002.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
