

New algorithms and an in silico benchmark for computational enzyme design
- PMID: 17132862
- PMCID: PMC2242439
- DOI: 10.1110/ps.062353106
New algorithms and an in silico benchmark for computational enzyme design
Abstract
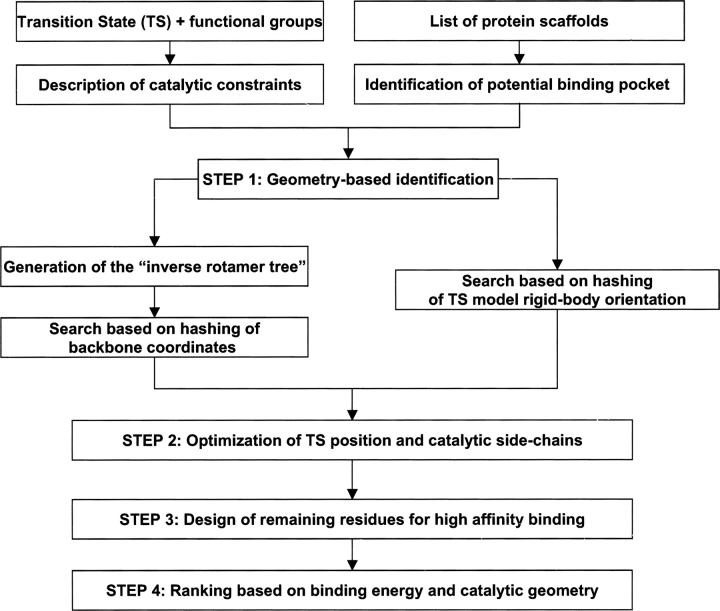
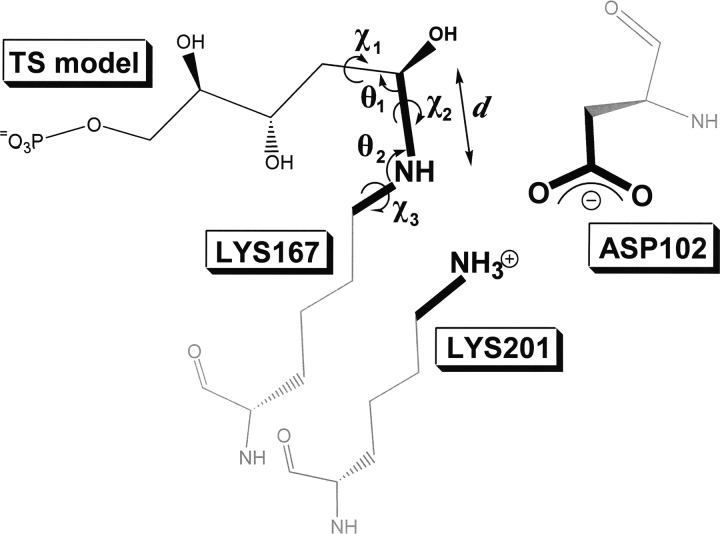
The creation of novel enzymes capable of catalyzing any desired chemical reaction is a grand challenge for computational protein design. Here we describe two new algorithms for enzyme design that employ hashing techniques to allow searching through large numbers of protein scaffolds for optimal catalytic site placement. We also describe an in silico benchmark, based on the recapitulation of the active sites of native enzymes, that allows rapid evaluation and testing of enzyme design methodologies. In the benchmark test, which consists of designing sites for each of 10 different chemical reactions in backbone scaffolds derived from 10 enzymes catalyzing the reactions, the new methods succeed in identifying the native site in the native scaffold and ranking it within the top five designs for six of the 10 reactions. The new methods can be directly applied to the design of new enzymes, and the benchmark provides a powerful in silico test for guiding improvements in computational enzyme design.
Figures




References
-
- Bachar, O., Fischer, D., Nussinov, R., and Wolfson, H. 1993. A computer vision based technique for 3-D sequence-independent structural comparison of proteins. Protein Eng. 6: 279–288. - PubMed
-
- Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., and Jain, S., et al. 2002. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 58: 899–907. - PubMed
-
- Clarke, N.D. and Yuan, S.M. 1995. Metal search: A computer program that helps design tetrahedral metal-binding sites. Proteins 23: 256–263. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
