

Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation
- PMID: 17143330
- PMCID: PMC1679711
- DOI: 10.1172/JCI29499
Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation
Abstract
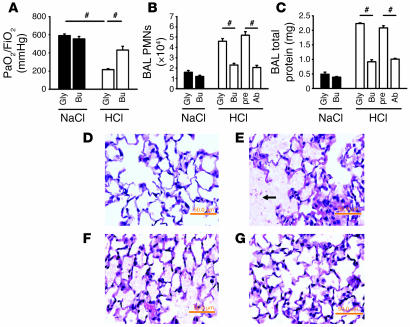
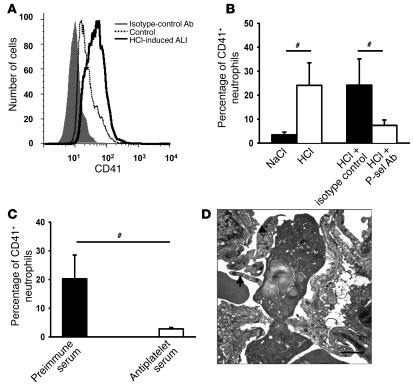
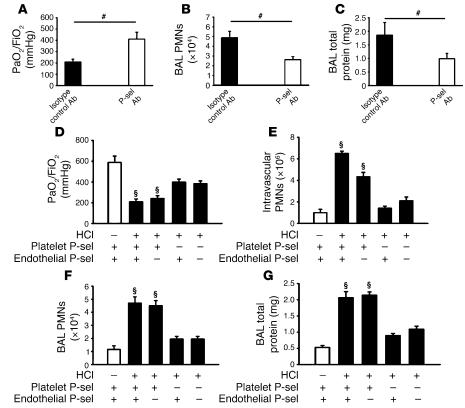
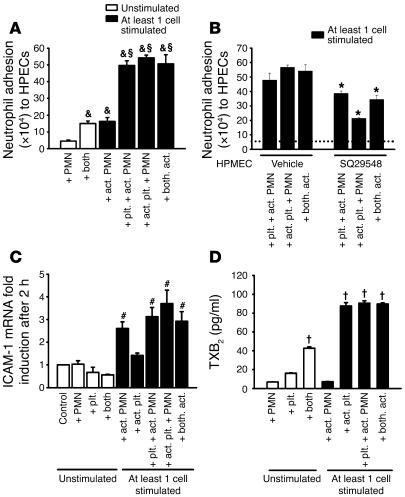
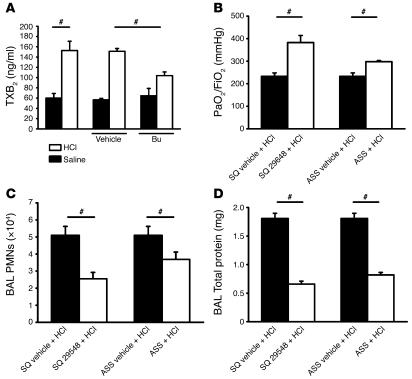
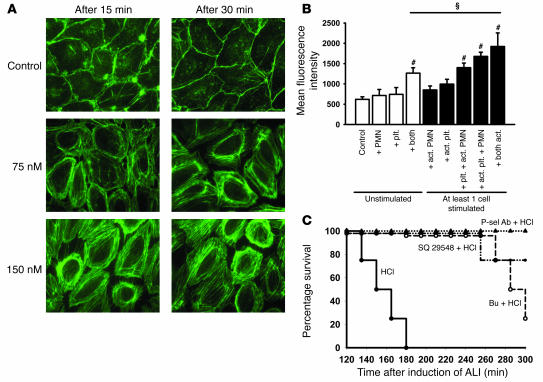
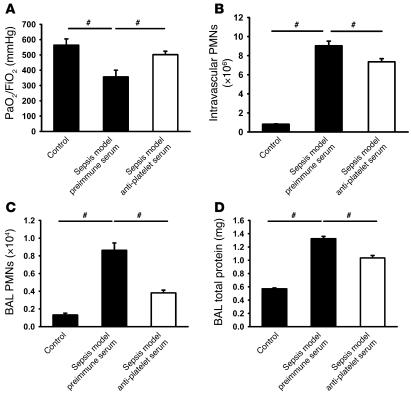
Acute lung injury (ALI) causes high mortality, but its molecular mechanisms are poorly understood. Acid aspiration is a frequent cause of ALI, leading to neutrophil sequestration, increased permeability, and deterioration of gas exchange. We investigated the role of platelet-neutrophil interactions in a murine model of acid-induced ALI. Acid aspiration induced P-selectin-dependent platelet-neutrophil interactions in blood and in lung capillaries. Reducing circulating platelets or blocking P-selectin halted the development of ALI. Bone marrow chimeras showed that platelet, not endothelial, P-selectin was responsible for the injury. The interaction of platelets with neutrophils and endothelia was associated with TXA(2) formation, with detrimental effects on permeability and tissue function. Activated platelets induced endothelial expression of ICAM-1 and increased neutrophil adhesion. Inhibition of platelet-neutrophil aggregation improved gas exchange, reduced neutrophil recruitment and permeability, and prolonged survival. The key findings were confirmed in a sepsis-induced model of ALI. These findings may translate into improved clinical treatments for ALI.
Figures
Comment in
-
Selectins revisited: the emerging role of platelets in inflammatory lung disease.J Clin Invest. 2006 Dec;116(12):3106-8. doi: 10.1172/JCI30664. J Clin Invest. 2006. PMID: 17143325 Free PMC article.
References
-
- Rubenfeld G.D., et al. Incidence and outcomes of acute lung injury. N. Engl. J. Med. 2005;353:1685–1693. - PubMed
-
- Warner M.A., Warner M.E., Weber J.G. Clinical significance of pulmonary aspiration during the perioperative period. Anesthesiology. 1993;78:56–62. - PubMed
-
- Olsson G.L., Hallen B., Hambraeus-Jonzon K. Aspiration during anaesthesia: a computer-aided study of 185,358 anaesthetics. Acta Anaesthesiol. Scand. 1986;30:84–92. - PubMed
-
- Marik P.E. Aspiration pneumonitis and aspiration pneumonia. N. Engl. J. Med. 2001;344:665–671. - PubMed
-
- Imai Y., et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. Jama. 2003;289:2104–2112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
