

Fast estimation of the difference between two PAM/JTT evolutionary distances in triplets of homologous sequences
- PMID: 17147817
- PMCID: PMC1762028
- DOI: 10.1186/1471-2105-7-529
Fast estimation of the difference between two PAM/JTT evolutionary distances in triplets of homologous sequences
Abstract
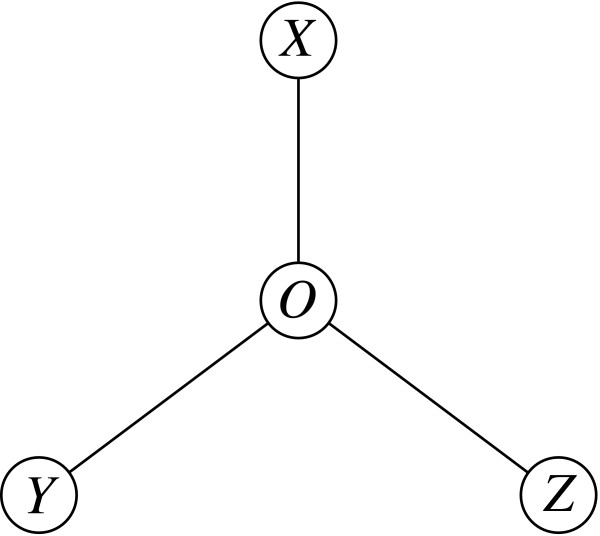
Background: The estimation of the difference between two evolutionary distances within a triplet of homologs is a common operation that is used for example to determine which of two sequences is closer to a third one. The most accurate method is currently maximum likelihood over the entire triplet. However, this approach is relatively time consuming.
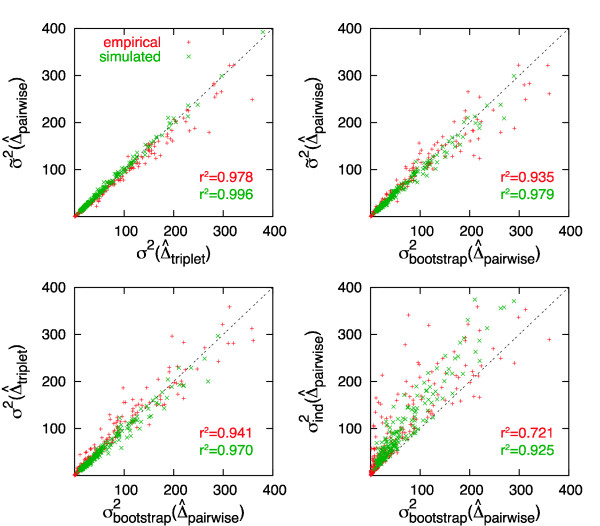
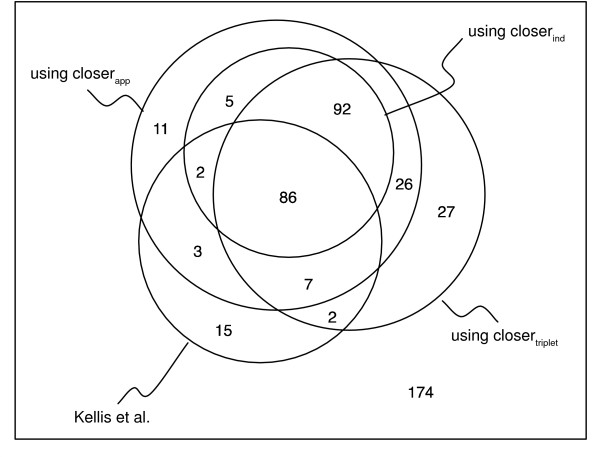
Results: We show that an alternative estimator, based on pairwise estimates and therefore much faster to compute, has almost the same statistical power as the maximum likelihood estimator. We also provide a numerical approximation for its variance, which could otherwise only be estimated through an expensive re-sampling approach such as bootstrapping. An extensive simulation demonstrates that the approximation delivers precise confidence intervals. To illustrate the possible applications of these results, we show how they improve the detection of asymmetric evolution, and the identification of the closest relative to a given sequence in a group of homologs.
Conclusion: The results presented in this paper constitute a basis for large-scale protein cross-comparisons of pairwise evolutionary distances.
Figures
Similar articles
-
Computing the all-pairs quartet distance on a set of evolutionary trees.J Bioinform Comput Biol. 2008 Feb;6(1):37-50. doi: 10.1142/s0219720008003266. J Bioinform Comput Biol. 2008. PMID: 18324744
-
Fast NJ-like algorithms to deal with incomplete distance matrices.BMC Bioinformatics. 2008 Mar 26;9:166. doi: 10.1186/1471-2105-9-166. BMC Bioinformatics. 2008. PMID: 18366787 Free PMC article.
-
Getting a tree fast: Neighbor Joining, FastME, and distance-based methods.Curr Protoc Bioinformatics. 2006 Oct;Chapter 6:Unit 6.3. doi: 10.1002/0471250953.bi0603s15. Curr Protoc Bioinformatics. 2006. PMID: 18428768
-
Homology assessment and molecular sequence alignment.J Biomed Inform. 2006 Feb;39(1):18-33. doi: 10.1016/j.jbi.2005.11.005. Epub 2005 Dec 9. J Biomed Inform. 2006. PMID: 16380300 Review.
-
Linkage disequilibrium for different scales and applications.Brief Bioinform. 2004 Dec;5(4):355-64. doi: 10.1093/bib/5.4.355. Brief Bioinform. 2004. PMID: 15606972 Review.
Cited by
-
Orthologous Matrix (OMA) algorithm 2.0: more robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference.Bioinformatics. 2017 Jul 15;33(14):i75-i82. doi: 10.1093/bioinformatics/btx229. Bioinformatics. 2017. PMID: 28881964 Free PMC article.
-
Algorithm of OMA for large-scale orthology inference.BMC Bioinformatics. 2008 Dec 4;9:518. doi: 10.1186/1471-2105-9-518. BMC Bioinformatics. 2008. PMID: 19055798 Free PMC article.
-
Controversies in modern evolutionary biology: the imperative for error detection and quality control.BMC Genomics. 2012 Jan 4;13:5. doi: 10.1186/1471-2164-13-5. BMC Genomics. 2012. PMID: 22217008 Free PMC article.
-
Covariance of maximum likelihood evolutionary distances between sequences aligned pairwise.BMC Evol Biol. 2008 Jun 23;8:179. doi: 10.1186/1471-2148-8-179. BMC Evol Biol. 2008. PMID: 18573206 Free PMC article.
-
Fast and accurate estimation of the covariance between pairwise maximum likelihood distances.PeerJ. 2014 Sep 25;2:e583. doi: 10.7717/peerj.583. eCollection 2014. PeerJ. 2014. PMID: 25279263 Free PMC article.
References
-
- Swofford DL, Olsen GL, Waddell PJ, Hillis DM. Phylogenetic inference. 2. Sunderland, Massachusetts: Sinauer Associates; 1996. pp. 407–514.
-
- Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. University of Washington. Seattle., Department of Genome Sciences; 2004.
-
- Dessimoz C, Cannarozzi G, Gil M, Margadant D, Roth A, Schneider A, Gonnet G. In: RECOMB 2005 Workshop on Comparative Genomics, Volume LNBI 3678 of Lecture Notes in Bioinformatics. McLysath A, Huson DH, editor. Springer-Verlag; 2005. OMA, A Comprehensive, Automated Project for the Identification of Orthologs from Complete Genome Data: Introduction and First Achievements; pp. 61–72.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
