

Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer's brain
- PMID: 17151277
- PMCID: PMC6674827
- DOI: 10.1523/JNEUROSCI.0575-06.2006
Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer's brain
Abstract
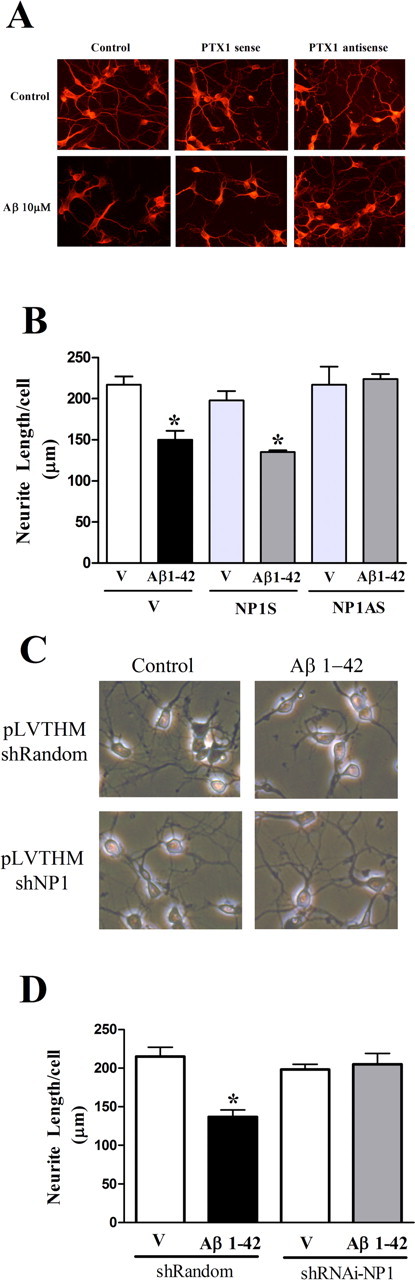
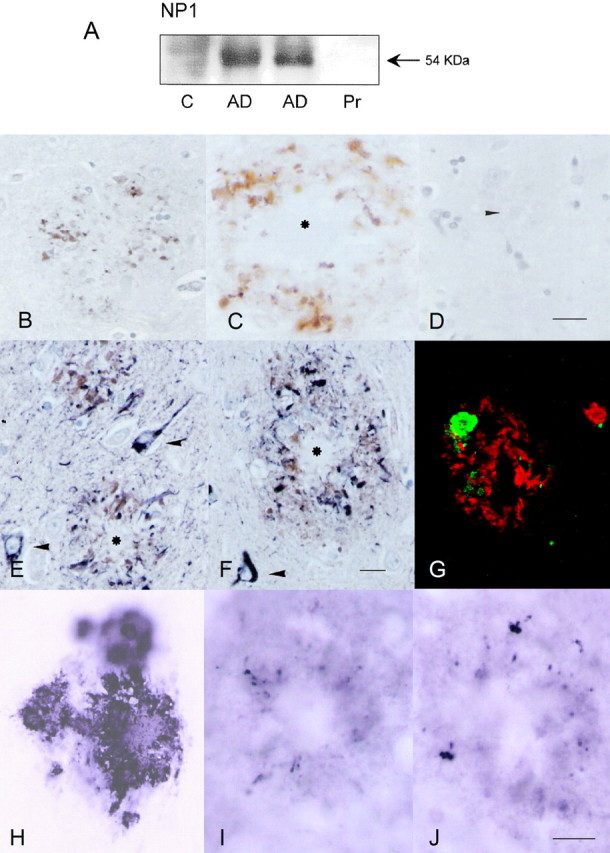
Accumulation of amyloid-beta (Abeta) is thought to play a central role in the progressive loss of synapses, the neurite damage, and the neuronal death that are characteristic in brains affected by Alzheimer's disease. However, the mechanisms through which Abeta produces such neurotoxicity remain unclear. Because Abeta depresses synaptic activity, we investigated whether the neurotoxicity of Abeta depends on the expression of NP1, a protein involved in excitatory synapse remodeling that has recently been shown to mediate neuronal death induced by reduction in neuronal activity in mature neurons. We found that treatment of cortical neurons in culture with Abeta produces a marked increase in NP1 protein that precedes apoptotic neurotoxicity. Silencing NP1 gene expression by RNA interference (short hairpin RNA for RNA interference) prevents the loss of synapses, the reduction in neurite outgrowth, and the apoptosis evoked by Abeta. Transgene overexpression of NP1 reproduced these neurotoxic effects of Abeta. Moreover, we found that NP1 was increased in dystrophic neurites of brains from patients with sporadic late-onset Alzheimer's disease. Dual immunohistochemistry for NP1 and tau showed that NP1 colocalizes with tau deposits in dystrophic neurites. Furthermore, NP1 colocalized with SNAP-25 (synaptosomal-associated protein of 25 kDa) in the majority of dystrophic neurites surrounding amyloid deposits. NP1 was also increased in cell processes surrounding amyloid plaques in the cerebral cortex and hippocampus of APP/PS1 (mutant amyloid precursor protein/presenilin 1) transgenic mice. These findings show that NP1 is a key factor for the synapse loss, the neurite damage, and the apoptotic neuronal death evoked by Abeta and indicate that Abeta contributes to the pathology of Alzheimer's disease by regulating NP1 expression.
Figures






References
-
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. - PubMed
-
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem. 2003;278:45937–45945. - PubMed
-
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89:1313–1317. - PubMed
-
- Bjartmar L, Huberman AD, Ullian EM, Renteria RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, Perin MS. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–6281. - PMC - PubMed
-
- Braak H, Braak E. Temporal sequence of Alzheimer's disease-related pathology. In: Peters A, Morrison JH, editors. Cerebral cortex. Neurodegenerative and age-related changes in structure and function of cerebral cortex. New York: Kluwer Academic/Plenum; 2001. pp. 475–512.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials