

doi: 10.1101/gr.5890907.
Epub 2006 Dec 6.
Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA
Affiliations
- PMID: 17151342
- PMCID: PMC1716261
- DOI: 10.1101/gr.5890907
Item in Clipboard
Exploring genomic dark matter: a critical assessment of the performance of homology search methods on noncoding RNA
Genome Res.
2007 Jan.
Abstract
Homology search is one of the most ubiquitous bioinformatic tasks, yet it is unknown how effective the currently available tools are for identifying noncoding RNAs (ncRNAs). In this work, we use reliable ncRNA data sets to assess the effectiveness of methods such as BLAST, FASTA, HMMer, and Infernal. Surprisingly, the most popular homology search methods are often the least accurate. As a result, many studies have used inappropriate tools for their analyses. On the basis of our results, we suggest homology search strategies using the currently available tools and some directions for future development.
Figures
An overview of homology search methods. A Venn diagram illustrating an overview of the methods used in this study. Different methods are classified as heuristic, single sequence, profile HMM, stochastic context-free grammar (SCFG), and/or RNA specific.

A comparison of the accuracy and efficiencies of homology search methods showing only the highest-ranking parameter settings for each algorithm from Supplemental Table 1. These were NCBI-BLAST (W7, 65%), WU-BLAST (W3), FASTA, ParAlign (65%), SSEARCH, HMMer (2.3.2, local), SAM (3.5, local), ERPIN, Infernal (0.7, local), RaveNnA, RSEARCH, and RSmatch. (A,B) Boxplots of algorithm ranks for the 5 and 20 sequence subsets, respectively. The blue curves show the median sensitivity, the green curve the median specificity, and the red curve the median MCC for each of the 12 programs. These accuracy values were computed by sampling either 5 or 20 sequences from the reference databases; these were used as input(s) to each algorithm for screening both the reference and a shuffled database. (C) Boxplots of algorithm speeds in nucleotides per second. The red curve shows median initialization times for the different programs. The single sequence, profile HMM, and RNA methods are displayed in unshaded, dark shaded, and lightly shaded boxes, respectively.
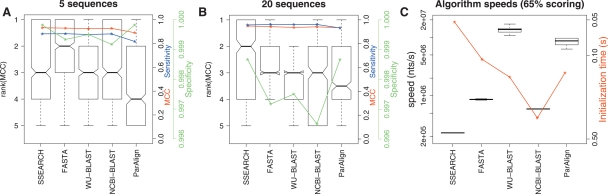
A comparison of the accuracy of sequence-based methods with the 65% scoring scheme and identical scoring parameters. These boxplots show the distributions of the ranks on MCC and timing data for each of the homology search methods when using a scoring scheme optimized for nucleotide sequences with 65% identity (match = +5, mismatch = −4, gapopen = 10, gapextension = 10). (A,B) Boxplots of algorithm ranks for the 5 and 20 sequence subsets, respectively. The blue curves show the median sensitivity, the green curve the median specificity, and the red curve the median MCC for each of the 12 programs. (C) Boxplots of algorithm speeds in nucleotides per second. The red curve shows median initialization times for the different programs.
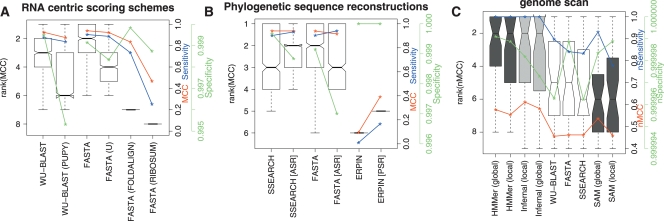
A comparison of the accuracy of methods using RNA-centric scoring matrices, phylogenetic sequence reconstructions, and the genome scan results. (A) A comparison of the accuracy of sequence-based methods with score matrices optimized for ncRNA. These boxplots show the distributions of the ranks on MCC for each of the homology search methods when using one of WU-BLAST (W7), WU-BLAST (W7, PUPY), FASTA, FASTA (U), FASTA (RIBOSUM), or FASTA (FOLDALIGN). These matrices are discussed in more detail in the text. (B) A comparison of FASTA, SSEARCH, and ERPIN with and without phylogenetic sequence reconstructions included in the input. Ancestral sequence reconstruction was used in the case of FASTA and SSEARCH and posterior predictive sequences in the case of ERPIN. Both A and B show results using 5 query sequences. (C) A set of representative programs from each category were run on human chromosome 12 (coordinates 90,000,000–130,000,000; ver NCBI35). The boxplot displays algorithm ranks; additionally, median nMCC, median nSensitivity, and median nSpecificity for each algorithm are displayed using the y-axis on the right. The single sequence, profile HMM, and RNA methods are displayed in unshaded, dark shaded, and lightly shaded boxes, respectively.
References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J., Gish W., Miller W., Myers E.W., Lipman D.J., Miller W., Myers E.W., Lipman D.J., Myers E.W., Lipman D.J., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Bollback J.P. Posterior mapping and predictive distributions. In: Nielsen R., editor. Statistical Methods in Molecular Evolution. Springer Verlag; New York: 2005. pp. 189–203.
-
- Cannone J., Subramanian S., Schnare M., Collett J., D’Souza L., Du Y., Feng B., Lin N., Madabusi L., Muller K., Subramanian S., Schnare M., Collett J., D’Souza L., Du Y., Feng B., Lin N., Madabusi L., Muller K., Schnare M., Collett J., D’Souza L., Du Y., Feng B., Lin N., Madabusi L., Muller K., Collett J., D’Souza L., Du Y., Feng B., Lin N., Madabusi L., Muller K., D’Souza L., Du Y., Feng B., Lin N., Madabusi L., Muller K., Du Y., Feng B., Lin N., Madabusi L., Muller K., Feng B., Lin N., Madabusi L., Muller K., Lin N., Madabusi L., Muller K., Madabusi L., Muller K., Muller K., et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials