

Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript
- PMID: 17157191
- PMCID: PMC1927835
- DOI: 10.1016/j.freeradbiomed.2006.09.014
Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript
Abstract
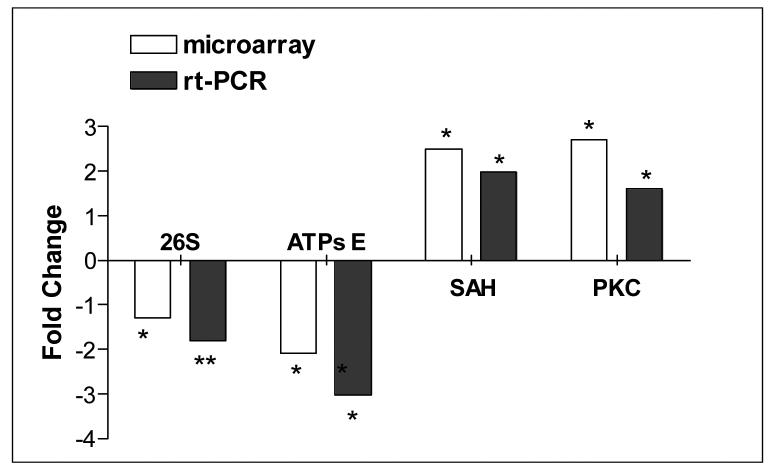
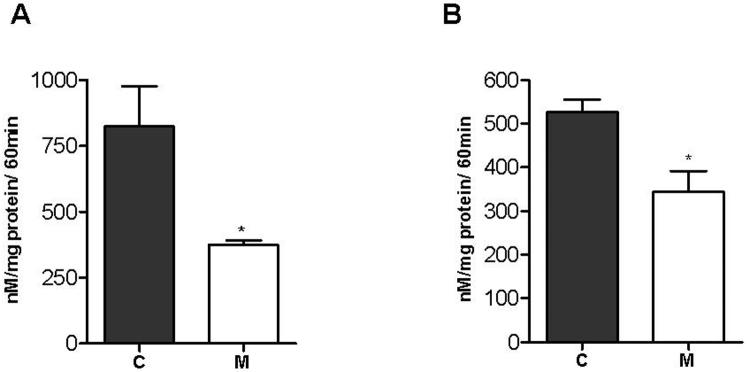
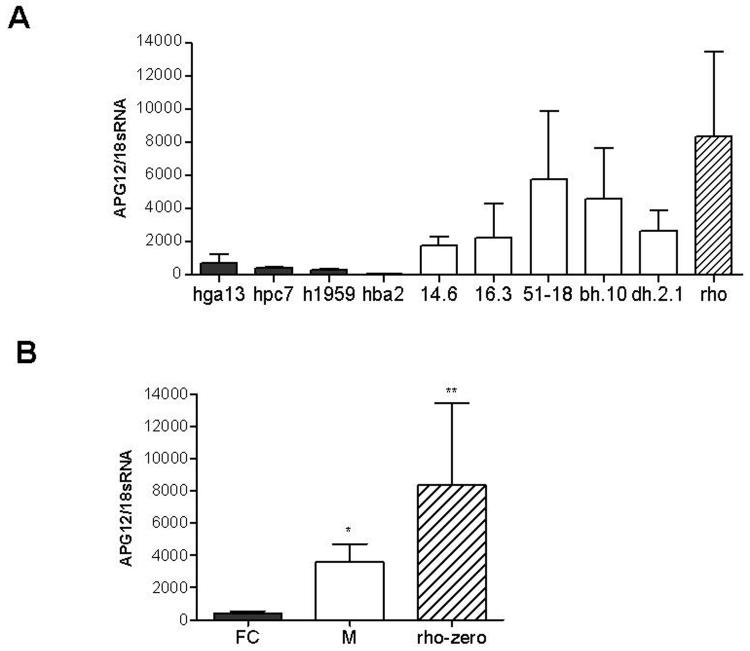
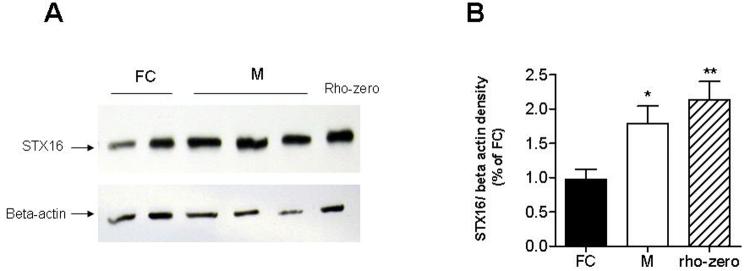
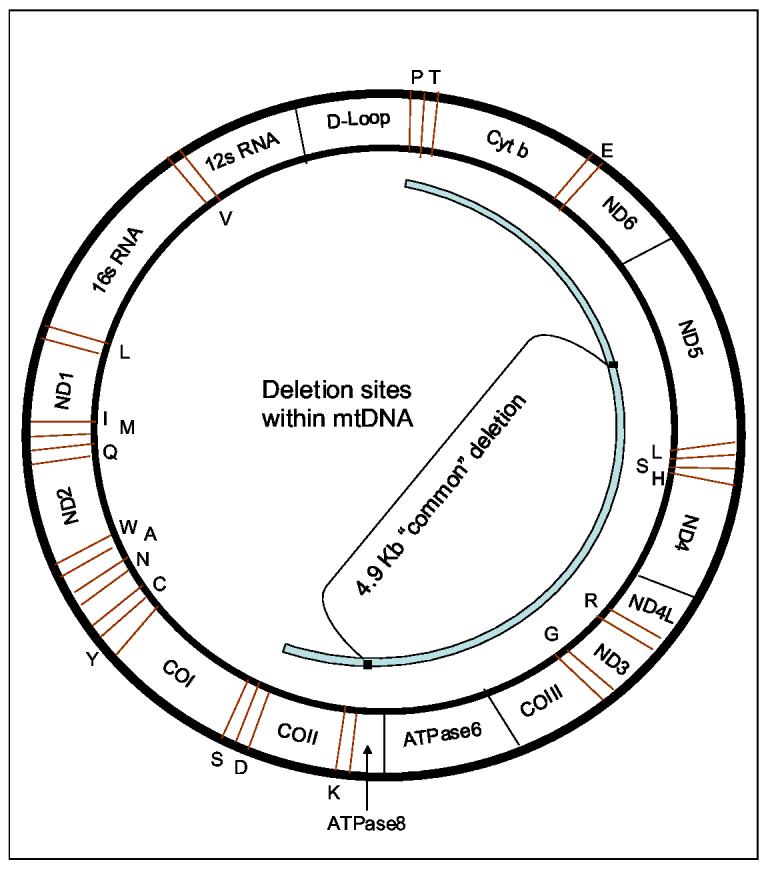
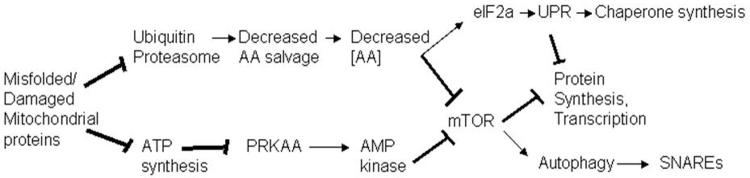
Deletions within the mitochondrial DNA (mtDNA) cause Kearns Sayre syndrome (KSS) and chronic progressive external opthalmoplegia (CPEO). The clinical signs of KSS include muscle weakness, heart block, pigmentary retinopathy, ataxia, deafness, short stature, and dementia. The identical deletions occur and rise exponentially as humans age, particularly in substantia nigra. Deletions at >30% concentration cause deficits in basic bioenergetic parameters, including membrane potential and ATP synthesis, but it is poorly understood how these alterations cause the pathologies observed in patients. To better understand the consequences of mtDNA deletions, we microarrayed six cell types containing mtDNA deletions from KSS and CPEO patients. There was a prominent inhibition of transcripts encoding ubiquitin-mediated proteasome activity, and a prominent induction of transcripts involved in the AMP kinase pathway, macroautophagy, and amino acid degradation. In mutant cells, we confirmed a decrease in proteasome biochemical activity, significantly lower concentration of several amino acids, and induction of an autophagic transcript. An interpretation consistent with the data is that mtDNA deletions increase protein damage, inhibit the ubiquitin-proteasome system, decrease amino acid salvage, and activate autophagy. This provides a novel pathophysiological mechanism for these diseases, and suggests potential therapeutic strategies.
Figures
Comment in
-
Mitochondrial DNA deletions and chloramphenicol treatment stimulate the autophagic transcript ATG12.Autophagy. 2007 Jul-Aug;3(4):377-80. doi: 10.4161/auto.4239. Epub 2007 Jul 5. Autophagy. 2007. PMID: 17457038
References
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. - PubMed
-
- Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, Nakase H, Bonilla E, Werneck LC, Servidei S, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N. Engl. J. Med. 1989;320:1293–1299. - PubMed
-
- Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science. 1989;244:346–349. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
