

Genome variation and evolution of the malaria parasite Plasmodium falciparum
- PMID: 17159978
- PMCID: PMC2663918
- DOI: 10.1038/ng1931
Genome variation and evolution of the malaria parasite Plasmodium falciparum
Erratum in
- Nat Genet. 2007 Apr;39(4):567
- Nat Genet. 2007 Mar;39(3):422
Abstract
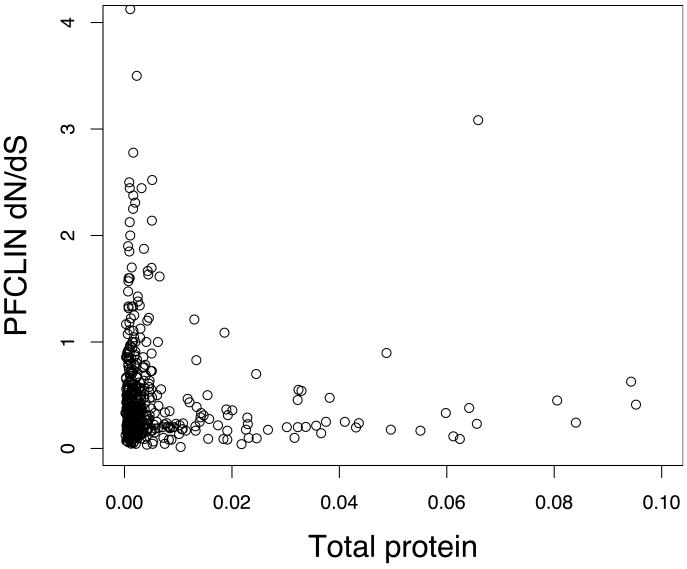
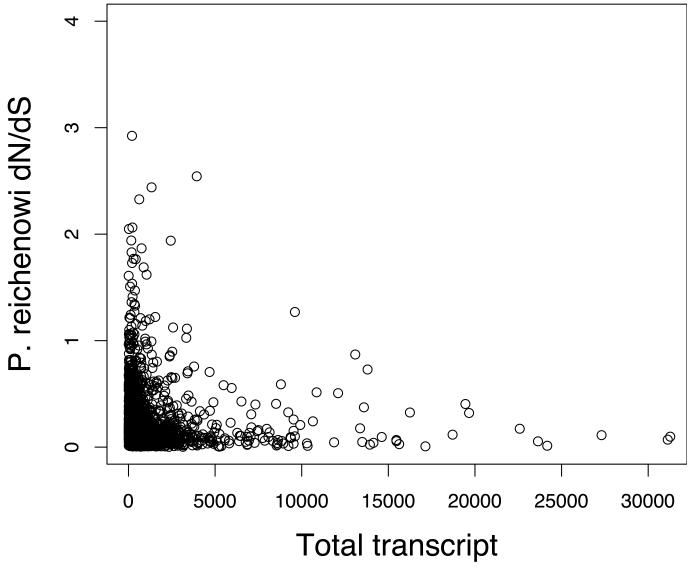
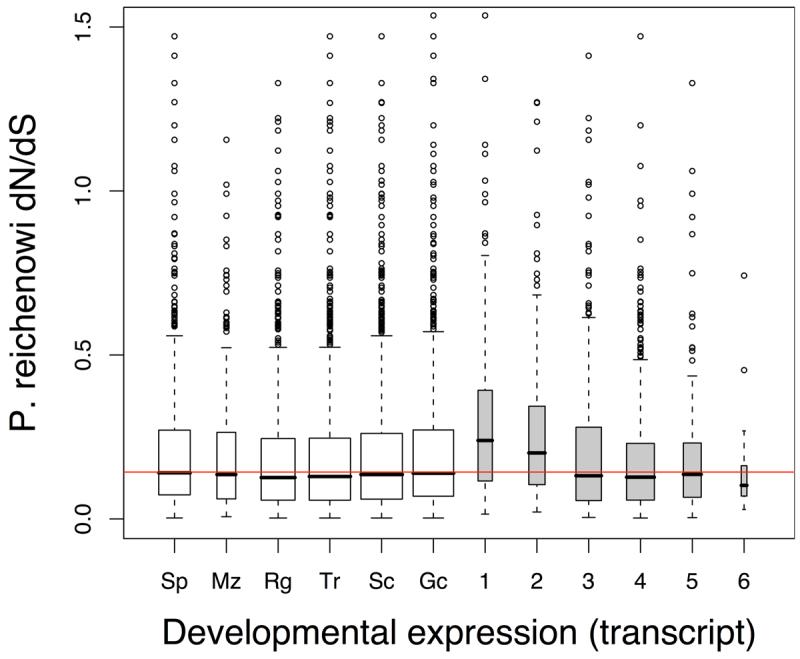
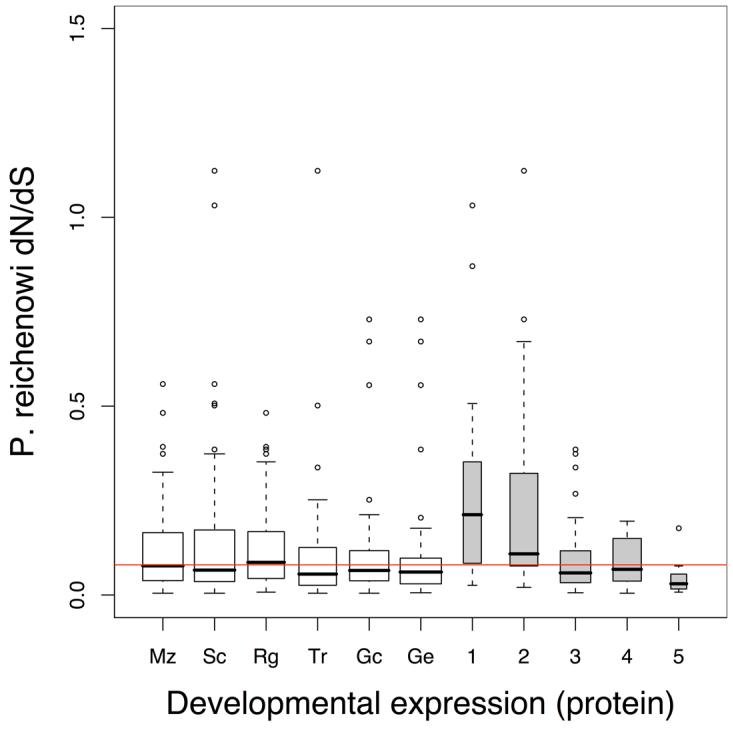
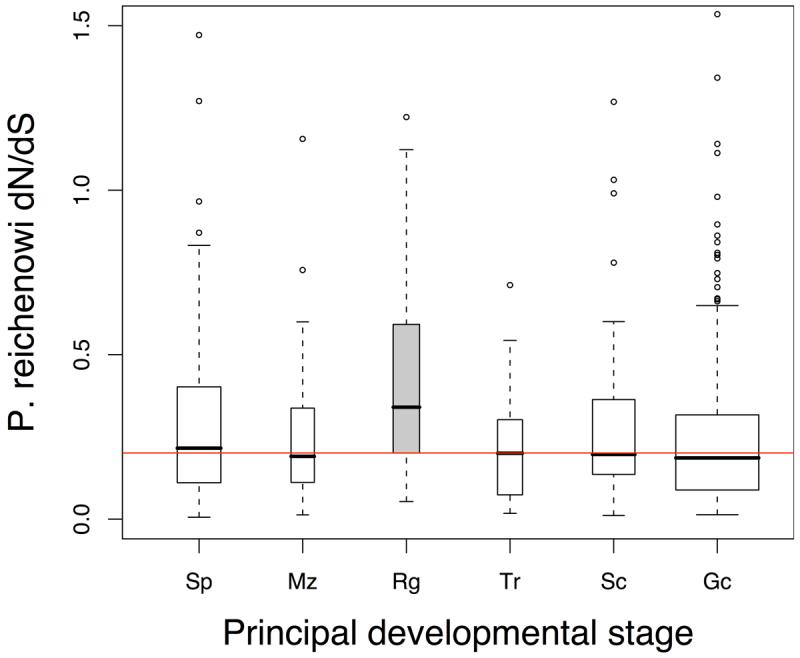
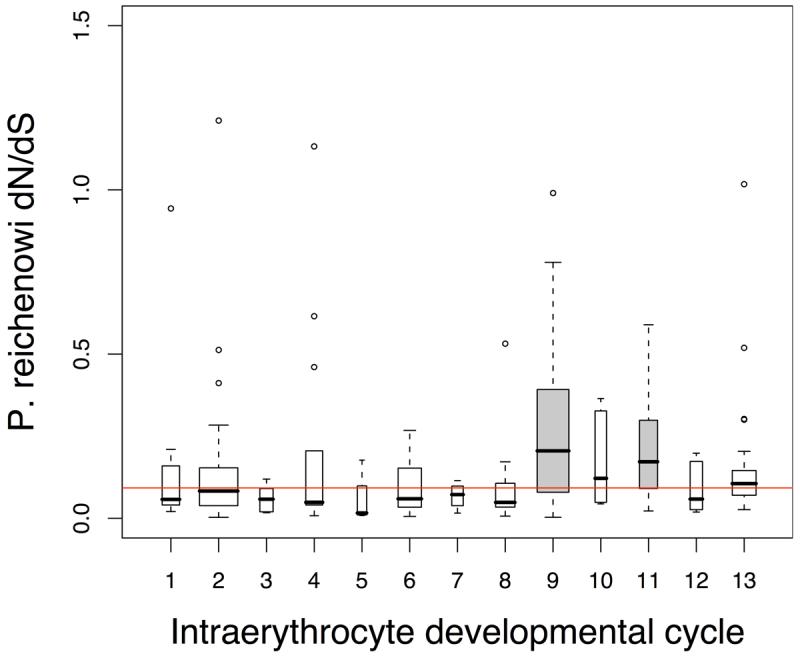
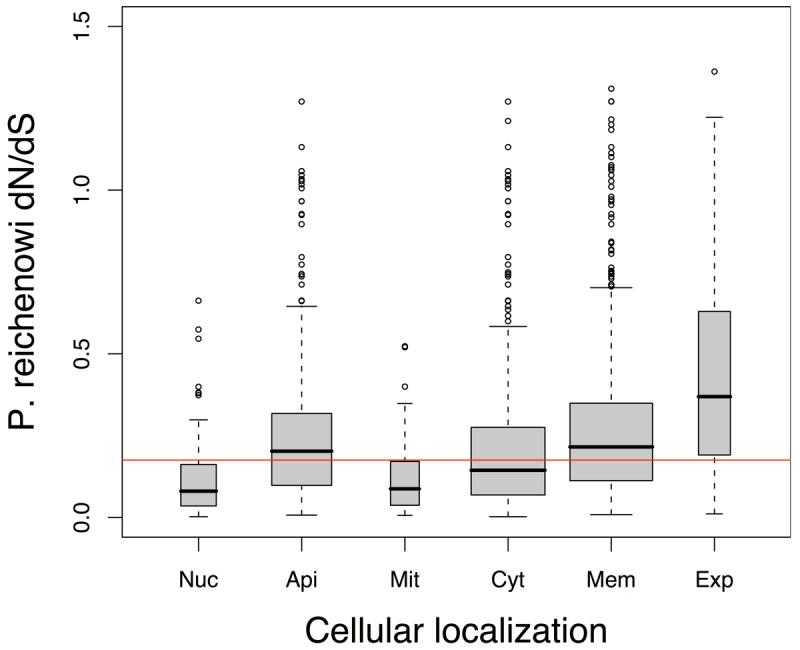
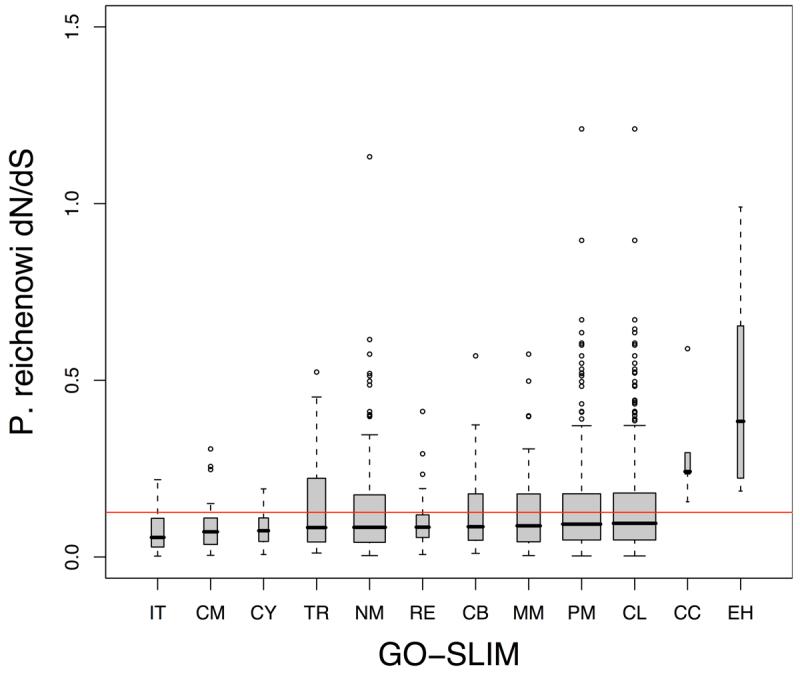
Infections with the malaria parasite Plasmodium falciparum result in more than 1 million deaths each year worldwide. Deciphering the evolutionary history and genetic variation of P. falciparum is critical for understanding the evolution of drug resistance, identifying potential vaccine candidates and appreciating the effect of parasite variation on prevalence and severity of malaria in humans. Most studies of natural variation in P. falciparum have been either in depth over small genomic regions (up to the size of a small chromosome) or genome wide but only at low resolution. In an effort to complement these studies with genome-wide data, we undertook shotgun sequencing of a Ghanaian clinical isolate (with fivefold coverage), the IT laboratory isolate (with onefold coverage) and the chimpanzee parasite P. reichenowi (with twofold coverage). We compared these sequences with the fully sequenced P. falciparum 3D7 isolate genome. We describe the most salient features of P. falciparum polymorphism and adaptive evolution with relation to gene function, transcript and protein expression and cellular localization. This analysis uncovers the primary evolutionary changes that have occurred since the P. falciparum-P. reichenowi speciation and changes that are occurring within P. falciparum.
Figures
Comment in
-
Toward a malaria haplotype map.Nat Genet. 2007 Jan;39(1):5-6. doi: 10.1038/ng0107-5. Nat Genet. 2007. PMID: 17192778 No abstract available.
References
-
- Korenromp E, Miller J, Nahlen B, Wardlaw T, Young M. World Malaria Report 2005. World Health Organization, Roll Back Malaria, Department and the United Nations Children's Fund. 2005
-
- Mu J, et al. Chromosome-wide SNPs reveal an ancient origin for Plasmodium falciparum. Nature. 2002;418:323–6. - PubMed
-
- Anderson TJ. Mapping drug resistance genes in Plasmodium falciparum by genome-wide association. Curr Drug Targets Infect Disord. 2004;4:65–78. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
