

Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia
- PMID: 17160902
- PMCID: PMC1785307
- DOI: 10.1086/510782
Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia
Abstract
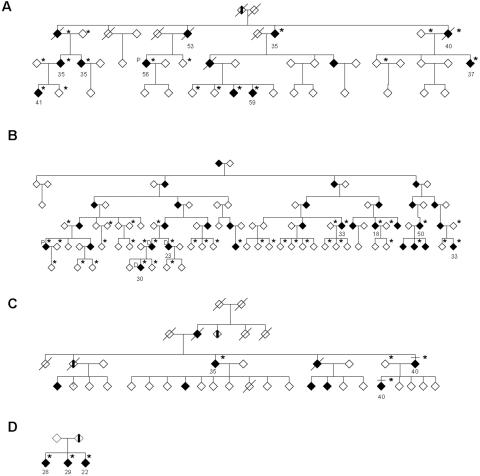
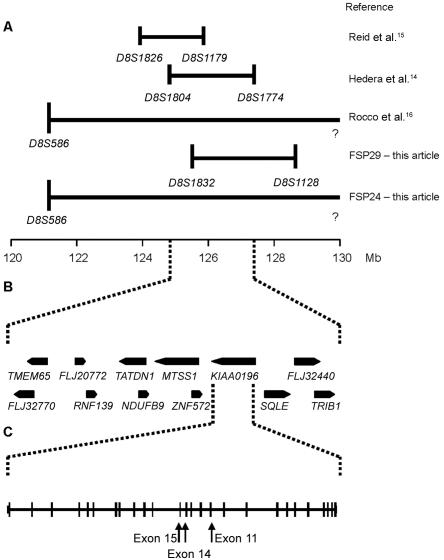
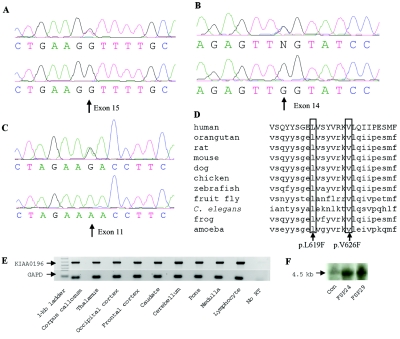
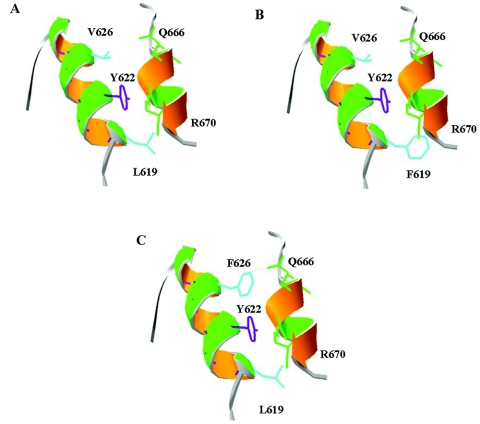
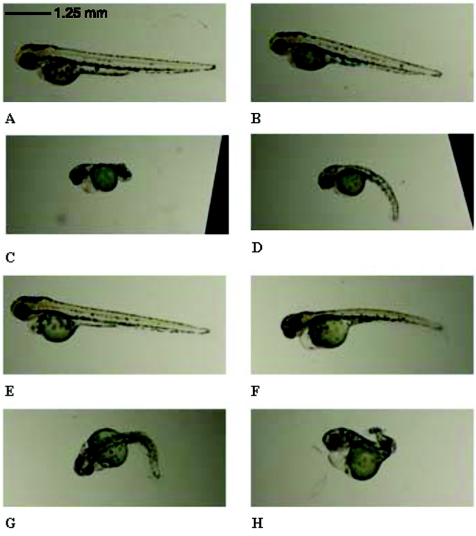
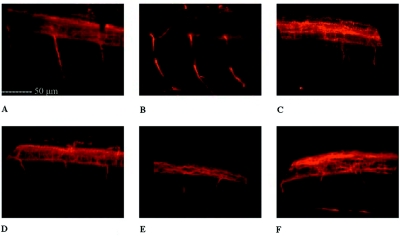
Hereditary spastic paraplegia (HSP) is a progressive upper-motor neurodegenerative disease. The eighth HSP locus, SPG8, is on chromosome 8p24.13. The three families previously linked to the SPG8 locus present with relatively severe, pure spastic paraplegia. We have identified three mutations in the KIAA0196 gene in six families that map to the SPG8 locus. One mutation, V626F, segregated in three large North American families with European ancestry and in one British family. An L619F mutation was found in a Brazilian family. The third mutation, N471D, was identified in a smaller family of European origin and lies in a spectrin domain. None of these mutations were identified in 500 control individuals. Both the L619 and V626 residues are strictly conserved across species and likely have a notable effect on the structure of the protein product strumpellin. Rescue studies with human mRNA injected in zebrafish treated with morpholino oligonucleotides to knock down the endogenous protein showed that mutations at these two residues impaired the normal function of the KIAA0196 gene. However, the function of the 1,159-aa strumpellin protein is relatively unknown. The identification and characterization of the KIAA0196 gene will enable further insight into the pathogenesis of HSP.
Figures
Similar articles
-
A novel strumpellin mutation and potential pitfalls in the molecular diagnosis of hereditary spastic paraplegia type SPG8.J Neurol Sci. 2014 Dec 15;347(1-2):372-4. doi: 10.1016/j.jns.2014.10.018. Epub 2014 Oct 16. J Neurol Sci. 2014. PMID: 25454649
-
Pure adult-onset spastic paraplegia caused by a novel mutation in the KIAA0196 (SPG8) gene.J Neurol. 2013 Jul;260(7):1765-9. doi: 10.1007/s00415-013-6870-x. Epub 2013 Mar 2. J Neurol. 2013. PMID: 23455931
-
The spectrum of KIAA0196 variants, and characterization of a murine knockout: implications for the mutational mechanism in hereditary spastic paraplegia type SPG8.Orphanet J Rare Dis. 2015 Nov 16;10:147. doi: 10.1186/s13023-015-0359-x. Orphanet J Rare Dis. 2015. PMID: 26572744 Free PMC article.
-
SPG8 mutations in Italian families: clinical data and literature review.Neurol Sci. 2020 Mar;41(3):699-703. doi: 10.1007/s10072-019-04180-z. Epub 2019 Dec 9. Neurol Sci. 2020. PMID: 31814071 Review.
-
Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms.Acta Neuropathol. 2013 Sep;126(3):307-28. doi: 10.1007/s00401-013-1115-8. Epub 2013 Jul 30. Acta Neuropathol. 2013. PMID: 23897027 Free PMC article. Review.
Cited by
-
Structural and functional studies of TBC1D23 C-terminal domain provide a link between endosomal trafficking and PCH.Proc Natl Acad Sci U S A. 2019 Nov 5;116(45):22598-22608. doi: 10.1073/pnas.1909316116. Epub 2019 Oct 17. Proc Natl Acad Sci U S A. 2019. PMID: 31624125 Free PMC article.
-
Physiology and pathology of endosome-to-Golgi retrograde sorting.Traffic. 2011 Aug;12(8):948-55. doi: 10.1111/j.1600-0854.2011.01188.x. Epub 2011 Apr 8. Traffic. 2011. PMID: 21382144 Free PMC article. Review.
-
Exome sequencing expands the mutational spectrum of SPG8 in a family with spasticity responsive to L-DOPA treatment.J Neurol. 2013 Sep;260(9):2414-6. doi: 10.1007/s00415-013-7044-6. Epub 2013 Jul 24. J Neurol. 2013. PMID: 23881105 Free PMC article. No abstract available.
-
Expression of N471D strumpellin leads to defects in the endolysosomal system.Dis Model Mech. 2018 Sep 13;11(9):dmm033449. doi: 10.1242/dmm.033449. Dis Model Mech. 2018. PMID: 30061306 Free PMC article.
-
Transcriptional and post-transcriptional regulation of SPAST, the gene most frequently mutated in hereditary spastic paraplegia.PLoS One. 2012;7(5):e36505. doi: 10.1371/journal.pone.0036505. Epub 2012 May 4. PLoS One. 2012. PMID: 22574173 Free PMC article.
References
Web Resources
-
- Conserved Domain Database, http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
-
- Fondation Jean Dausset–CEPH, http://www.cephb.fr/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for C. familiaris [accession number XP_532327], P. troglodytes [accession number XP_519952], D. melanogaster [accession number CG12272], C. elegans [accession number CE13235], X. tropicalis [accession number MGC89323], R. norvegicus [accession number XP_343250], D. rerio [accession number BC045490], G. gallus [accession number XP_418441], D. discoideum [accession number EAL63144], M. musculus [accession number NP_705776.2], and H. sapiens mRNA [accession number NM_014846.2])
-
- InterPro, http://www.ebi.ac.uk/interpro/ (for InterProScan from the European Bioinformatics Institute)
-
- NCBI BLAST, http://www.ncbi.nlm.nih.gov/blast/
References
-
- Polo JM, Calleja J, Combarros O, Berciano J (1991) Hereditary ataxias and paraplegias in Cantabria, Spain: an epidemiological and clinical study. Brain 114:855–866 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
