

The congenital muscular dystrophies: recent advances and molecular insights
- PMID: 17163796
- PMCID: PMC2855646
- DOI: 10.2350/06-07-0127.1
The congenital muscular dystrophies: recent advances and molecular insights
Abstract
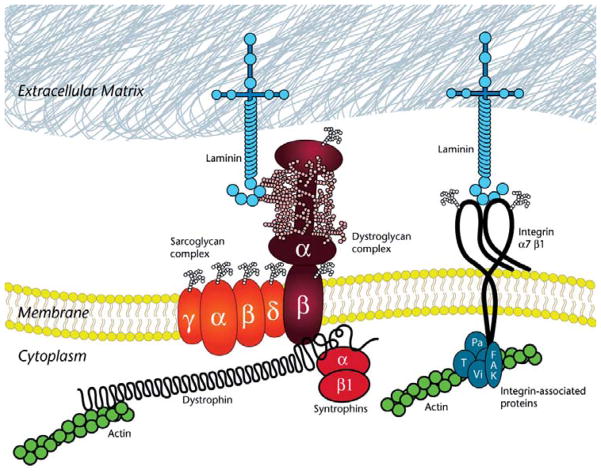
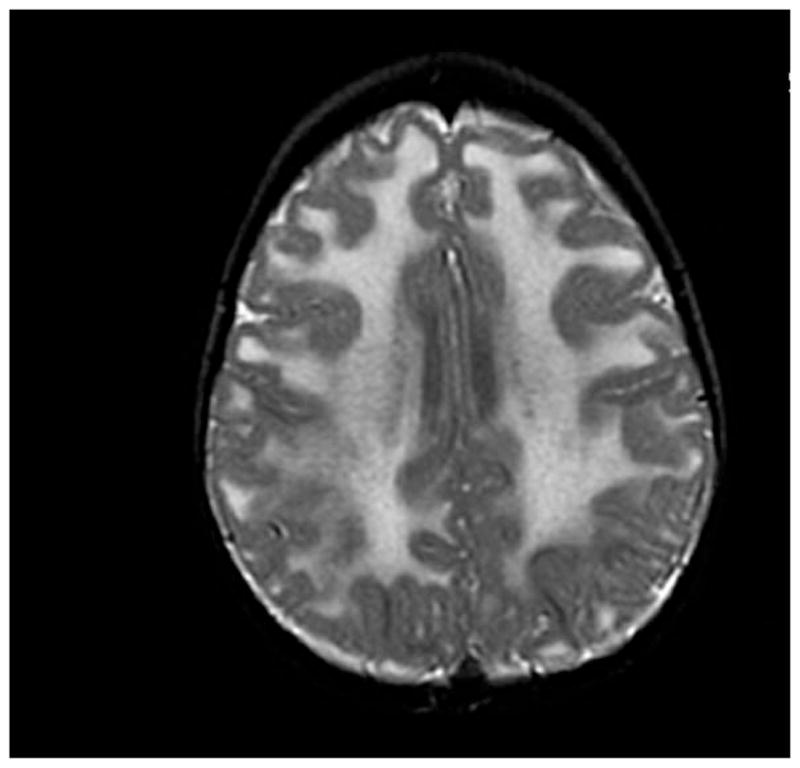
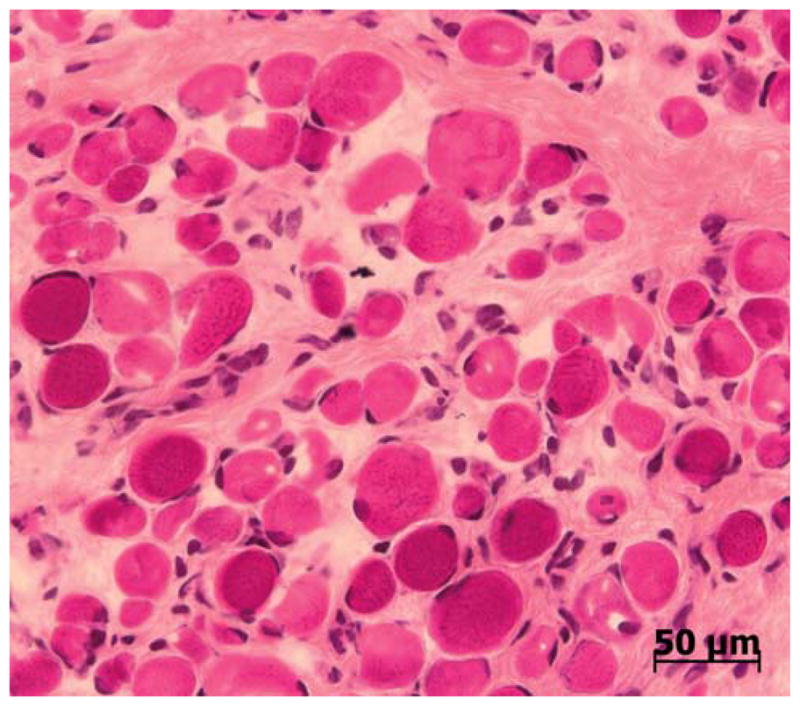
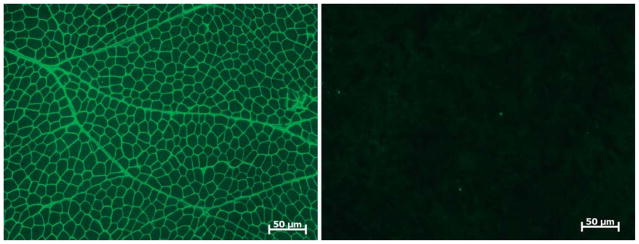
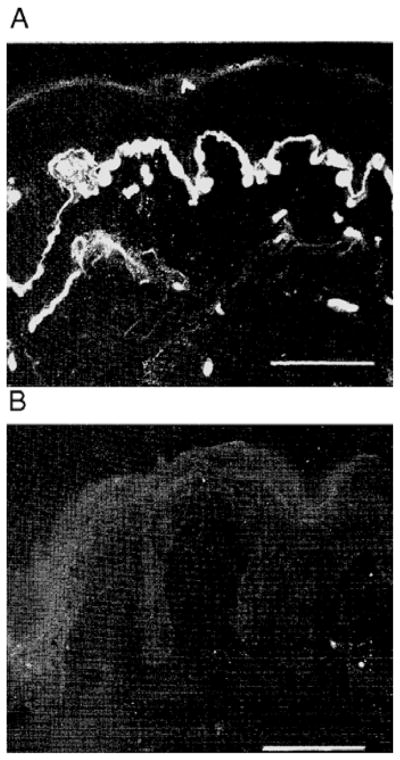
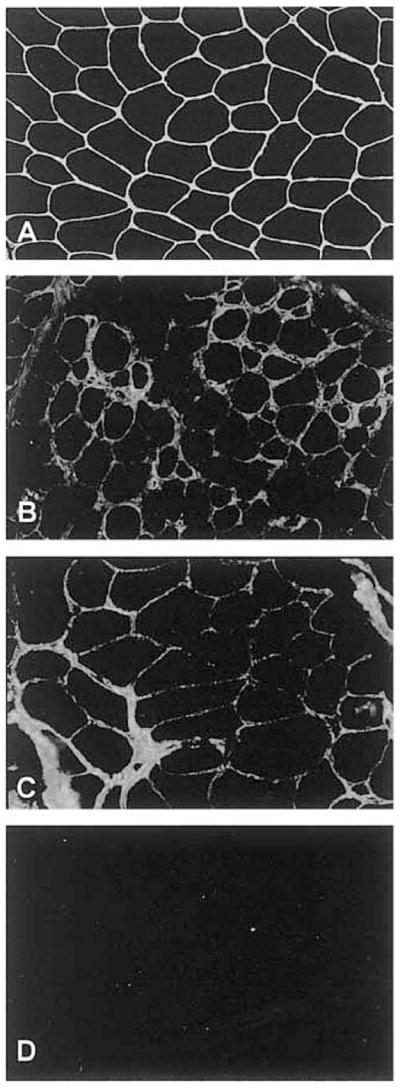
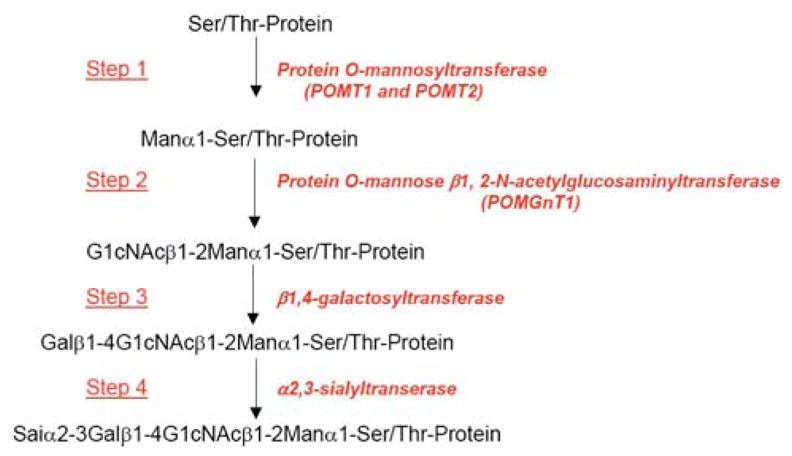
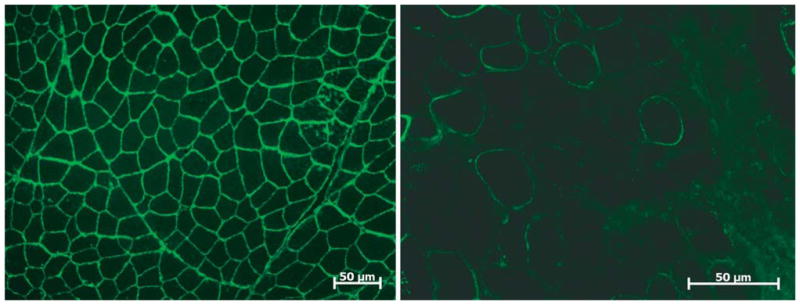
Over the past decade, molecular understanding of the congenital muscular dystrophies (CMDs) has greatly expanded. The diseases can be classified into 3 major groups based on the affected genes and the location of their expressed protein: abnormalities of extracellular matrix proteins (LAMA2, COL6A1, COL6A2, COL6A3), abnormalities of membrane receptors for the extracellular matrix (fukutin, POMGnT1, POMT1, POMT2, FKRP, LARGE, and ITGA7), and abnormal endoplasmic reticulum protein (SEPN1). The diseases begin in the perinatal period or shortly thereafter. A specific diagnosis can be challenging because the muscle pathology is usually not distinctive. Immunostaining of muscle using a battery of antibodies can help define a disorder that will need confirmation by gene testing. In muscle diseases with overlapping pathological features, such as CMD, careful attention to the clinical clues (e.g., family history, central nervous system features) can help guide the battery of immunostains necessary to target an unequivocal diagnosis.
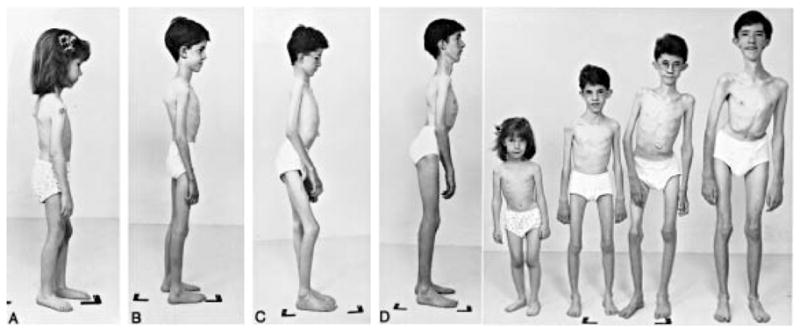
Figures
Similar articles
-
Congenital Muscular Dystrophy Overview – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY.2001 Jan 22 [updated 2012 Aug 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2001 Jan 22 [updated 2012 Aug 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301468 Free Books & Documents. Review.
-
Diagnosis and etiology of congenital muscular dystrophy.Neurology. 2008 Jul 29;71(5):312-21. doi: 10.1212/01.wnl.0000284605.27654.5a. Epub 2007 Dec 26. Neurology. 2008. PMID: 18160674
-
Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan.Am J Hum Genet. 2001 Dec;69(6):1198-209. doi: 10.1086/324412. Epub 2001 Oct 8. Am J Hum Genet. 2001. PMID: 11592034 Free PMC article.
-
Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan.Brain. 2007 Oct;130(Pt 10):2725-35. doi: 10.1093/brain/awm212. Epub 2007 Sep 18. Brain. 2007. PMID: 17878207
-
[Congenital muscular dystrophies in children].Rev Neurol. 2013 Sep 6;57 Suppl 1:S47-52. Rev Neurol. 2013. PMID: 23897156 Review. Spanish.
Cited by
-
Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat.Mol Med. 2013 May 20;19(1):79-87. doi: 10.2119/molmed.2013.00011. Mol Med. 2013. PMID: 23552722 Free PMC article.
-
Elevated expression of the integrin-associated protein PINCH suppresses the defects of Drosophila melanogaster muscle hypercontraction mutants.PLoS Genet. 2013 Mar;9(3):e1003406. doi: 10.1371/journal.pgen.1003406. Epub 2013 Mar 28. PLoS Genet. 2013. PMID: 23555310 Free PMC article.
-
Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle.J Proteome Res. 2012 Sep 7;11(9):4413-24. doi: 10.1021/pr300328r. Epub 2012 Jul 30. J Proteome Res. 2012. PMID: 22775139 Free PMC article.
-
Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases.Mol Med. 2011 May-Jun;17(5-6):457-65. doi: 10.2119/molmed.2011.00049. Epub 2011 Feb 7. Mol Med. 2011. PMID: 21308150 Free PMC article. Review.
-
Multifaceted antibodies development against synthetic α-dystroglycan mucin glycopeptide as promising tools for dystroglycanopathies diagnostic.Glycoconj J. 2020 Feb;37(1):77-93. doi: 10.1007/s10719-019-09893-z. Epub 2019 Dec 10. Glycoconj J. 2020. PMID: 31823246
References
-
- Engvall E. Laminin variants: why, where, and when? Kidney Int. 1993;43:2–6. - PubMed
-
- Muntoni F, Voit T. The congenital dystrophies in 2004: a century of exciting progress. Neuromuscul Disord. 2004;14:635–649. - PubMed
-
- Philpot J, Sewry C, Pennock J, Dubowitz Clnical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromusc Disord. 1995;5:301–305. - PubMed
-
- Fardeau M, Tome FM, Helfling-Leclerc A, et al. Congenital muscular dystrophy with merosin deficiency: clinical, histopathological, immunocytochemical and genetic analysis. Rev Neurol. 1996;152:11–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
