

Insights into the evolutionary history of tubercle bacilli as disclosed by genetic rearrangements within a PE_PGRS duplicated gene pair
- PMID: 17163995
- PMCID: PMC1762029
- DOI: 10.1186/1471-2148-6-107
Insights into the evolutionary history of tubercle bacilli as disclosed by genetic rearrangements within a PE_PGRS duplicated gene pair
Abstract
Background: The highly homologous PE_PGRS (Proline-glutamic acid_polymorphic GC-rich repetitive sequence) genes are members of the PE multigene family which is found only in mycobacteria. PE genes are particularly abundant within the genomes of pathogenic mycobacteria where they seem to have expanded as a result of gene duplication events. PE_PGRS genes are characterized by their high GC content and extensive repetitive sequences, making them prone to recombination events and genetic variability.
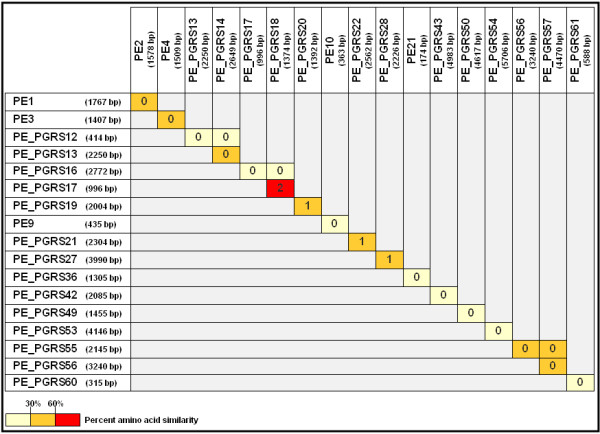
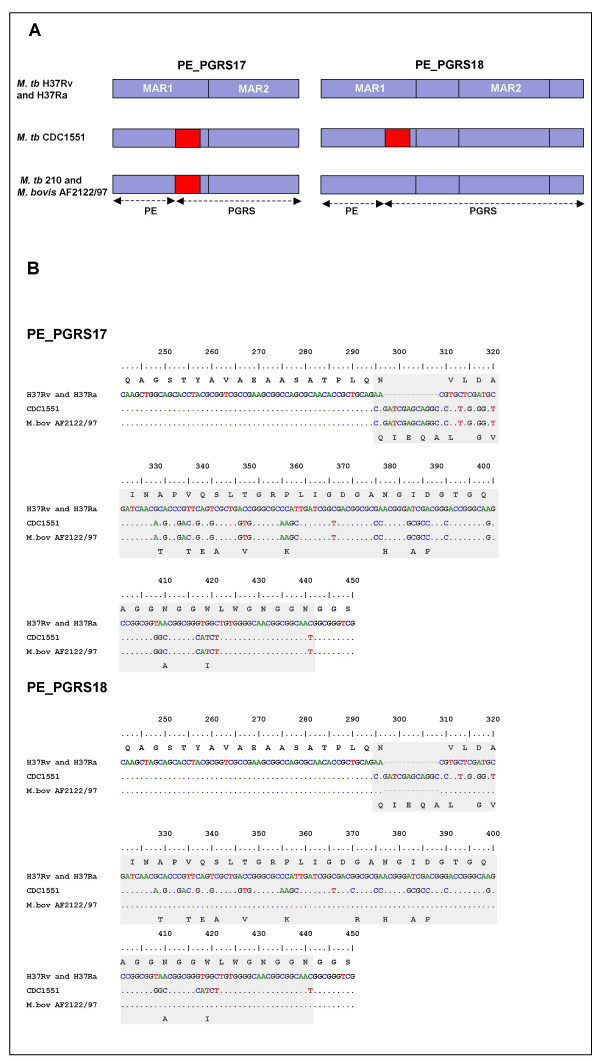
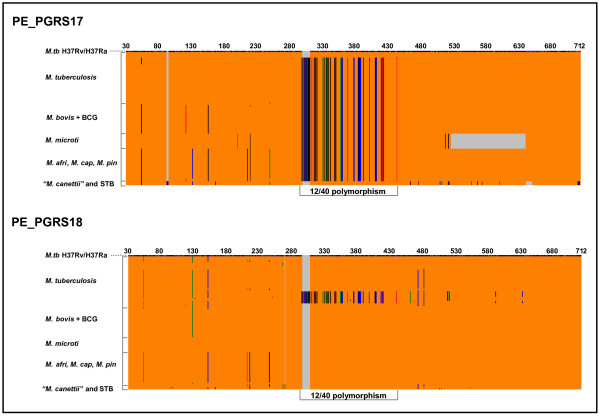
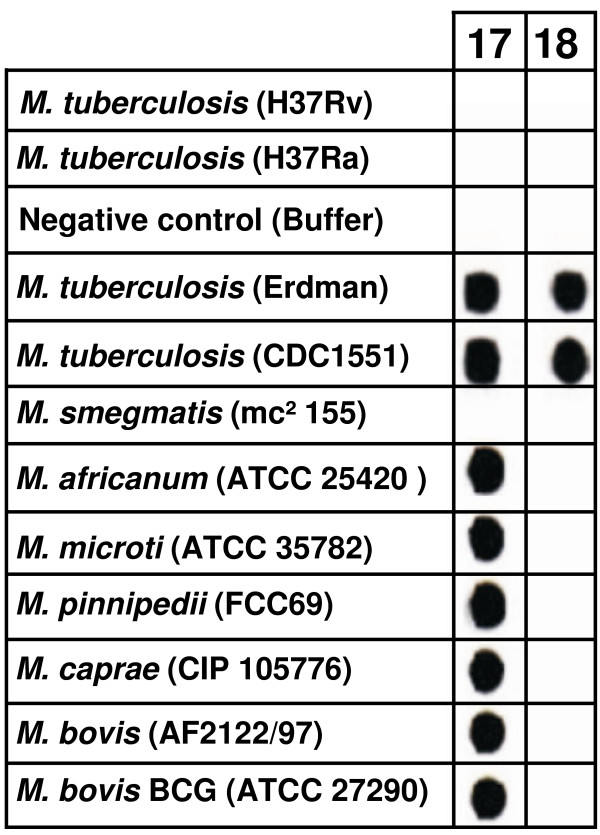
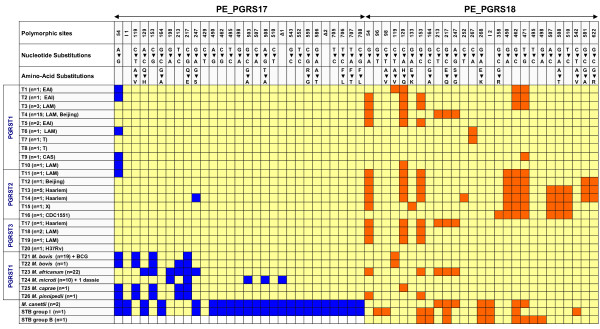
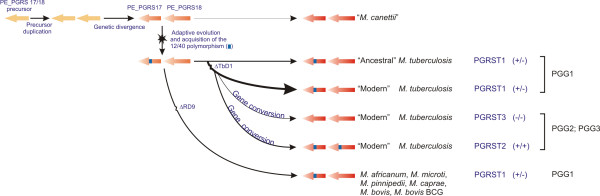
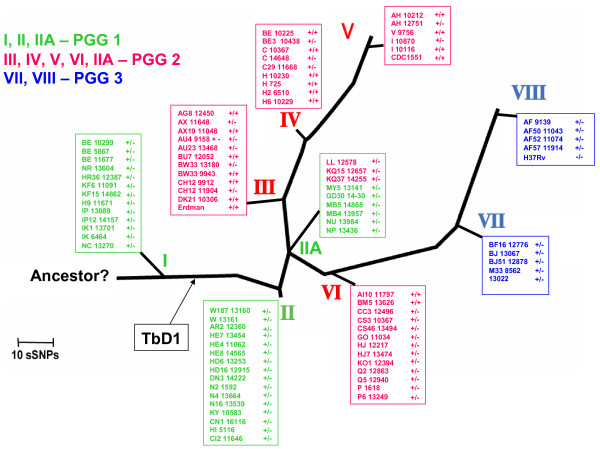
Results: Comparative sequence analysis of Mycobacterium tuberculosis genes PE_PGRS17 (Rv0978c) and PE_PGRS18 (Rv0980c) revealed a striking genetic variation associated with this typical tandem duplicate. In comparison to the M. tuberculosis reference strain H37Rv, the variation (named the 12/40 polymorphism) consists of an in-frame 12-bp insertion invariably accompanied by a set of 40 single nucleotide polymorphisms (SNPs) that occurs either in PE_PGRS17 or in both genes. Sequence analysis of the paralogous genes in a representative set of worldwide distributed tubercle bacilli isolates revealed data which supported previously proposed evolutionary scenarios for the M. tuberculosis complex (MTBC) and confirmed the very ancient origin of "M. canettii" and other smooth tubercle bacilli. Strikingly, the identified polymorphism appears to be coincident with the emergence of the post-bottleneck successful clone from which the MTBC expanded. Furthermore, the findings provide direct and clear evidence for the natural occurrence of gene conversion in mycobacteria, which appears to be restricted to modern M. tuberculosis strains.
Conclusion: This study provides a new perspective to explore the molecular events that accompanied the evolution, clonal expansion, and recent diversification of tubercle bacilli.
Figures


Similar articles
-
Evolution of smooth tubercle Bacilli PE and PE_PGRS genes: evidence for a prominent role of recombination and imprint of positive selection.PLoS One. 2013 May 21;8(5):e64718. doi: 10.1371/journal.pone.0064718. Print 2013. PLoS One. 2013. PMID: 23705005 Free PMC article.
-
Variation of the Mycobacterium tuberculosis PE_PGRS 33 gene among clinical isolates.J Clin Microbiol. 2005 Oct;43(10):4954-60. doi: 10.1128/JCM.43.10.4954-4960.2005. J Clin Microbiol. 2005. PMID: 16207947 Free PMC article.
-
An overview to understand the role of PE_PGRS family proteins in Mycobacterium tuberculosis H37 Rv and their potential as new drug targets.Biotechnol Appl Biochem. 2015 Mar-Apr;62(2):145-53. doi: 10.1002/bab.1266. Epub 2014 Nov 11. Biotechnol Appl Biochem. 2015. PMID: 24975480 Review.
-
Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions.BMC Evol Biol. 2006 Nov 15;6:95. doi: 10.1186/1471-2148-6-95. BMC Evol Biol. 2006. PMID: 17105670 Free PMC article.
-
PE and PPE Genes: A Tale of Conservation and Diversity.Adv Exp Med Biol. 2017;1019:191-207. doi: 10.1007/978-3-319-64371-7_10. Adv Exp Med Biol. 2017. PMID: 29116636 Review.
Cited by
-
Evolution of smooth tubercle Bacilli PE and PE_PGRS genes: evidence for a prominent role of recombination and imprint of positive selection.PLoS One. 2013 May 21;8(5):e64718. doi: 10.1371/journal.pone.0064718. Print 2013. PLoS One. 2013. PMID: 23705005 Free PMC article.
-
Evidence for a rapid rate of molecular evolution at the hypervariable and immunogenic Mycobacterium tuberculosis PPE38 gene region.BMC Evol Biol. 2009 Sep 21;9:237. doi: 10.1186/1471-2148-9-237. BMC Evol Biol. 2009. PMID: 19769792 Free PMC article.
-
Consequences of genomic diversity in Mycobacterium tuberculosis.Semin Immunol. 2014 Dec;26(6):431-44. doi: 10.1016/j.smim.2014.09.012. Epub 2014 Oct 22. Semin Immunol. 2014. PMID: 25453224 Free PMC article. Review.
-
PE_PGRS38 Interaction With HAUSP Downregulates Antimycobacterial Host Defense via TRAF6.Front Immunol. 2022 Apr 28;13:862628. doi: 10.3389/fimmu.2022.862628. eCollection 2022. Front Immunol. 2022. PMID: 35572598 Free PMC article.
-
HIV-1 replication is differentially regulated by distinct clinical strains of Mycobacterium tuberculosis.PLoS One. 2009 Jul 1;4(7):e6116. doi: 10.1371/journal.pone.0006116. PLoS One. 2009. PMID: 19568431 Free PMC article.
References
-
- World Health Organization . Global Tuberculosis Control Surveillance, Planning, Financing. World Health Organization, Geneva; 2005.
-
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Musser JM. Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA. 1997;94:9869–9874. doi: 10.1073/pnas.94.18.9869. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
