

High precision multi-genome scale reannotation of enzyme function by EFICAz
- PMID: 17166279
- PMCID: PMC1764738
- DOI: 10.1186/1471-2164-7-315
High precision multi-genome scale reannotation of enzyme function by EFICAz
Abstract
Background: The functional annotation of most genes in newly sequenced genomes is inferred from similarity to previously characterized sequences, an annotation strategy that often leads to erroneous assignments. We have performed a reannotation of 245 genomes using an updated version of EFICAz, a highly precise method for enzyme function prediction.
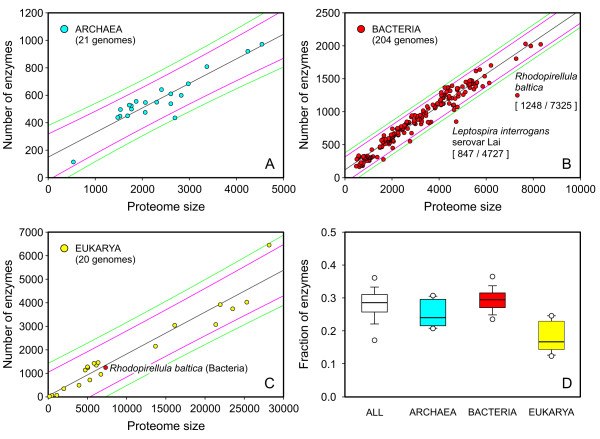
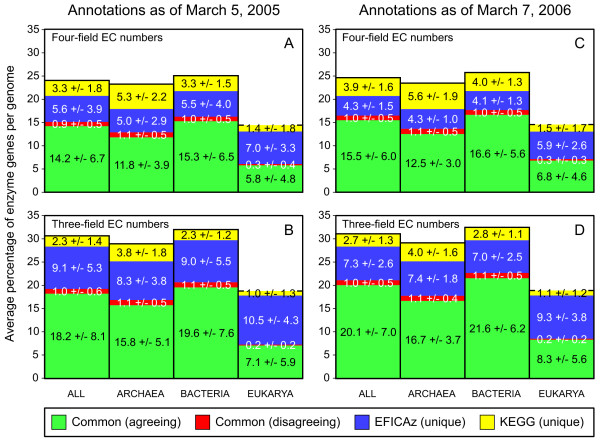
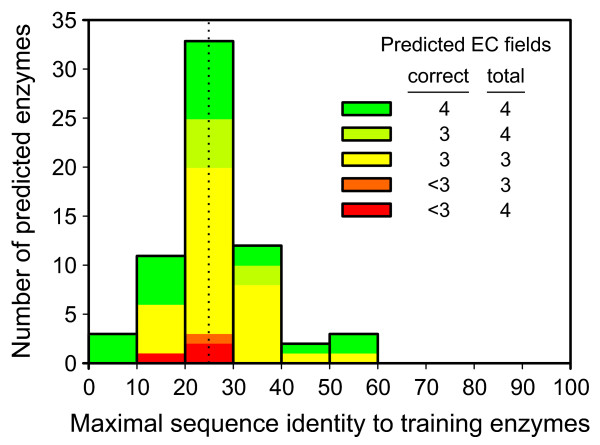
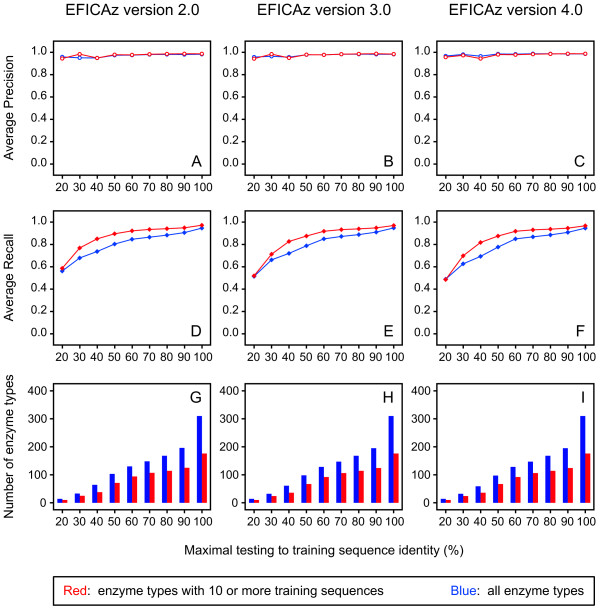
Results: Based on our three-field EC number predictions, we have obtained lower-bound estimates for the average enzyme content in Archaea (29%), Bacteria (30%) and Eukarya (18%). Most annotations added in KEGG from 2005 to 2006 agree with EFICAz predictions made in 2005. The coverage of EFICAz predictions is significantly higher than that of KEGG, especially for eukaryotes. Thousands of our novel predictions correspond to hypothetical proteins. We have identified a subset of 64 hypothetical proteins with low sequence identity to EFICAz training enzymes, whose biochemical functions have been recently characterized and find that in 96% (84%) of the cases we correctly identified their three-field (four-field) EC numbers. For two of the 64 hypothetical proteins: PA1167 from Pseudomonas aeruginosa, an alginate lyase (EC 4.2.2.3) and Rv1700 of Mycobacterium tuberculosis H37Rv, an ADP-ribose diphosphatase (EC 3.6.1.13), we have detected annotation lag of more than two years in databases. Two examples are presented where EFICAz predictions act as hypothesis generators for understanding the functional roles of hypothetical proteins: FLJ11151, a human protein overexpressed in cancer that EFICAz identifies as an endopolyphosphatase (EC 3.6.1.10), and MW0119, a protein of Staphylococcus aureus strain MW2 that we propose as candidate virulence factor based on its EFICAz predicted activity, sphingomyelin phosphodiesterase (EC 3.1.4.12).
Conclusion: Our results suggest that we have generated enzyme function annotations of high precision and recall. These predictions can be mined and correlated with other information sources to generate biologically significant hypotheses and can be useful for comparative genome analysis and automated metabolic pathway reconstruction.
Figures
References
-
- Friedberg I. Automated protein function prediction--the genomic challenge. Brief Bioinform. 2006 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
