

Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair
- PMID: 17169990
- PMCID: PMC1802609
- DOI: 10.1093/nar/gkl1069
Ascorbate acts as a highly potent inducer of chromate mutagenesis and clastogenesis: linkage to DNA breaks in G2 phase by mismatch repair
Abstract
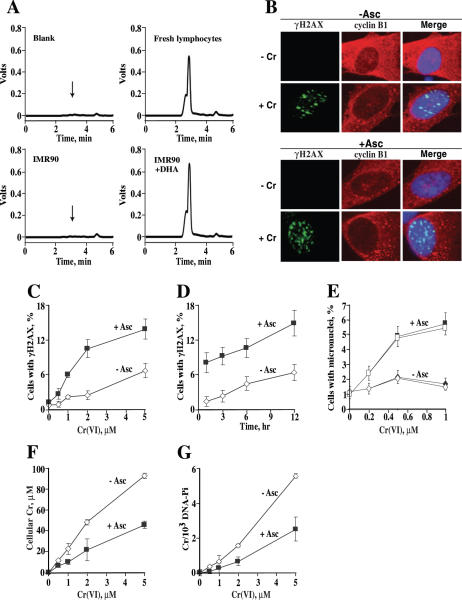
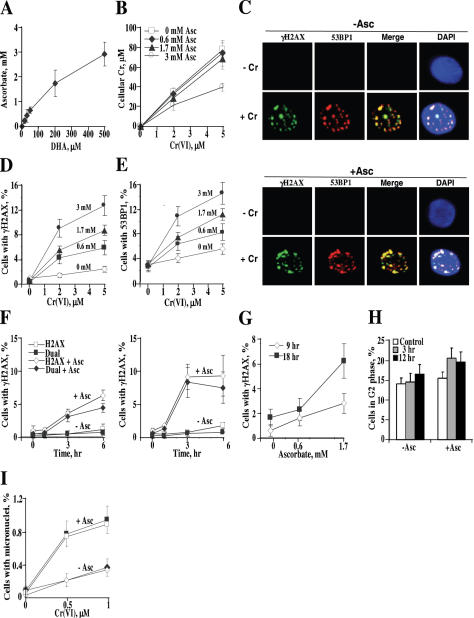
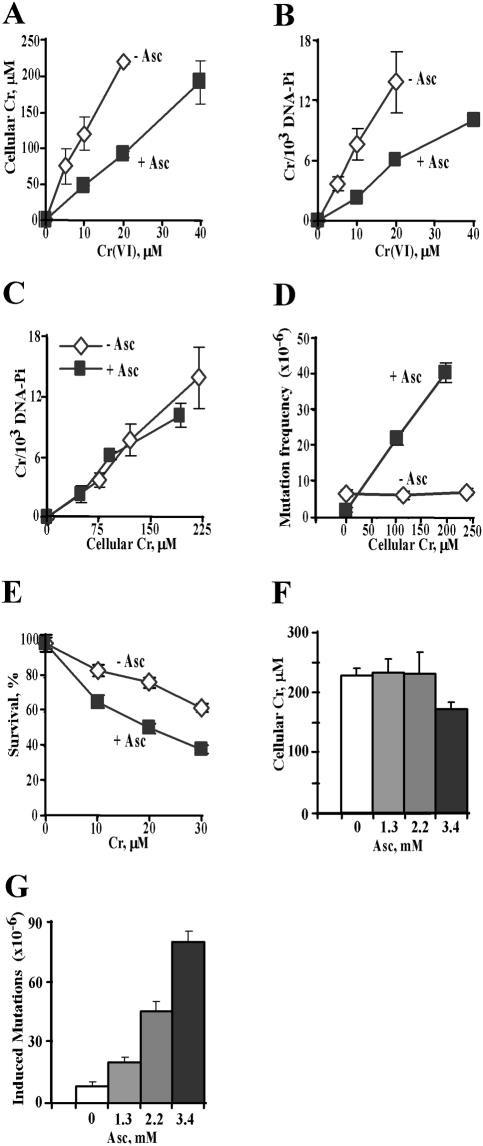
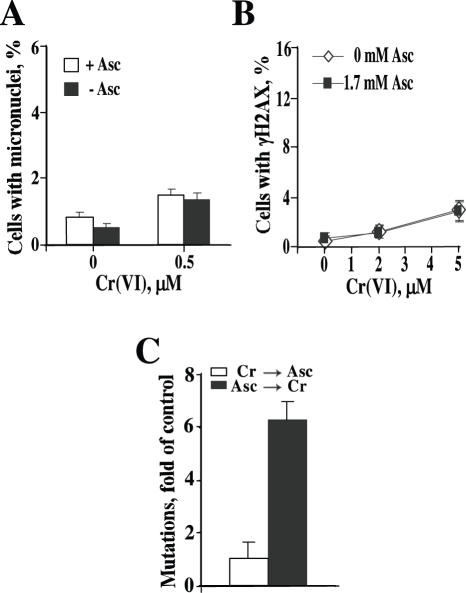
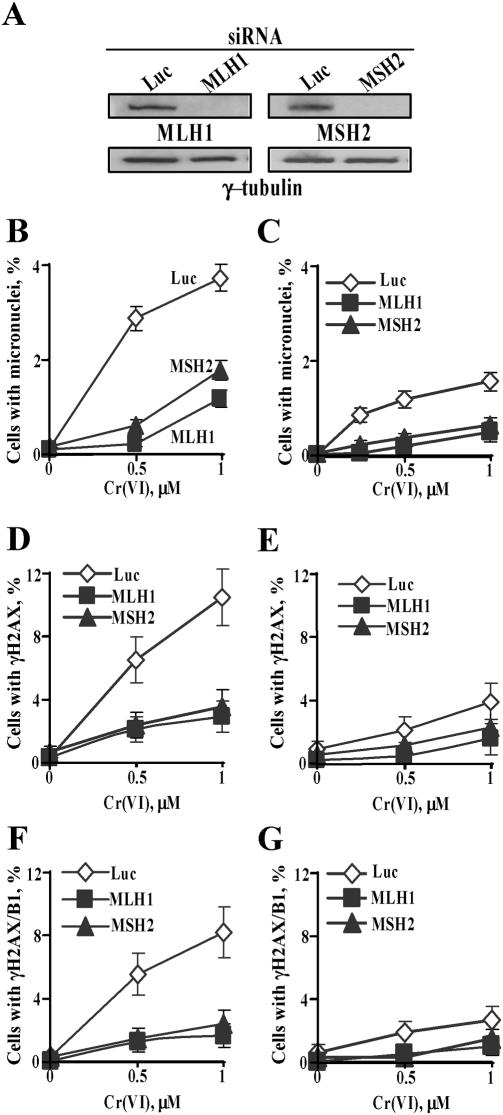
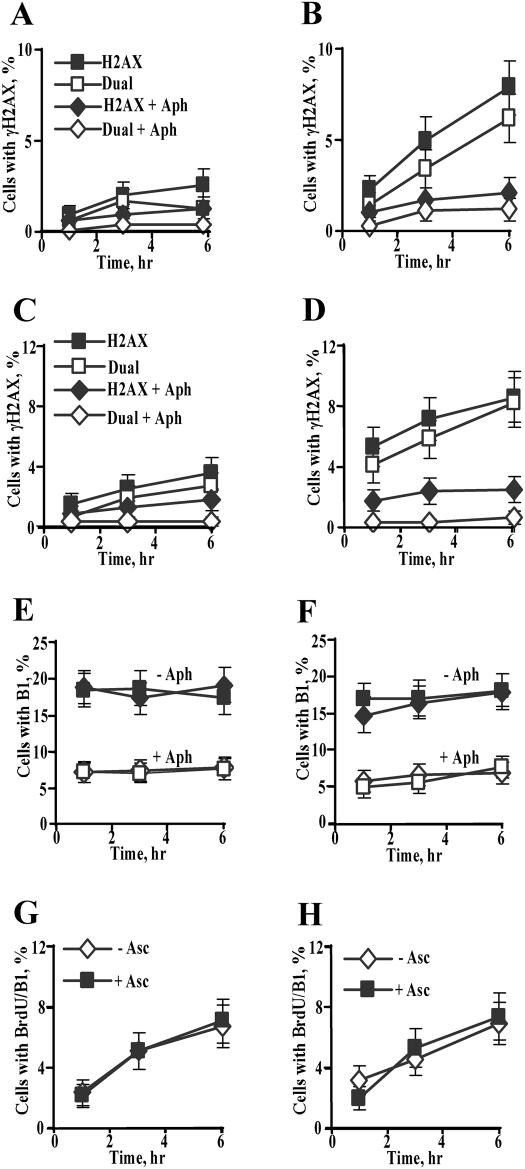
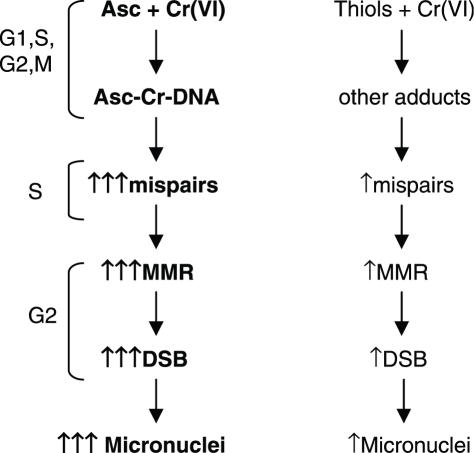
Here we examined the role of cellular vitamin C in genotoxicity of carcinogenic chromium(VI) that requires reduction to induce DNA damage. In the presence of ascorbate (Asc), low 0.2-2 microM doses of Cr(VI) caused 10-15 times more chromosomal breakage in primary human bronchial epithelial cells or lung fibroblasts. DNA double-strand breaks (DSB) were preferentially generated in G2 phase as detected by colocalization of H2AX and 53BP1 foci in cyclin B1-expressing cells. Asc dramatically increased the formation of centromere-negative micronuclei, demonstrating that induced DSB were inefficiently repaired. DSB in G2 cells were caused by aberrant mismatch repair of Cr damage in replicated DNA, as DNA polymerase inhibitor aphidicolin and silencing of MSH2 or MLH1 by shRNA suppressed induction of H2AX and micronuclei. Cr(VI) was also up to 10 times more mutagenic in cells containing Asc. Increasing Asc concentrations generated progressively more mutations and DSB, revealing the genotoxic potential of otherwise nontoxic Cr(VI) doses. Asc amplified genotoxicity of Cr(VI) by altering the spectrum of DNA damage, as total Cr-DNA binding was unchanged and post-Cr loading of Asc exhibited no effects. Collectively, these studies demonstrated that Asc-dependent metabolism is the main source of genotoxic and mutagenic damage in Cr(VI)-exposed cells.
Figures
References
-
- Langard S. One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am. J. Ind. Med. 1990;17:189–215. - PubMed
-
- Zhitkovich A. Chromium: exposure, toxicity and biomonitoring approaches. In: Wilson S.H., Suk W.A., editors. Biomarkers of Environmentally Associated Disease: Technologies, Concepts, and Perspective. New York: CRC Press LLC; 2002. pp. 269–287.
-
- De Flora S. Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis. 2000;21:533–541. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
