

Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain
- PMID: 17177598
- PMCID: PMC1702556
- DOI: 10.1371/journal.pmed.0030485
Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain
Abstract
Background: Protein tyrosine kinases are important regulators of cellular homeostasis with tightly controlled catalytic activity. Mutations in kinase-encoding genes can relieve the autoinhibitory constraints on kinase activity, can promote malignant transformation, and appear to be a major determinant of response to kinase inhibitor therapy. Missense mutations in the EGFR kinase domain, for example, have recently been identified in patients who showed clinical responses to EGFR kinase inhibitor therapy.
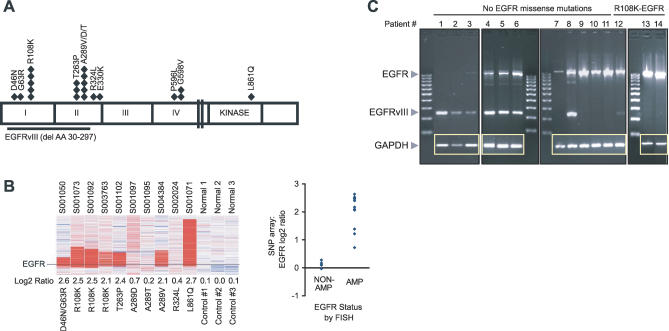
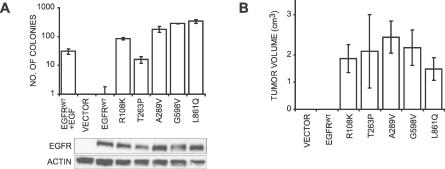
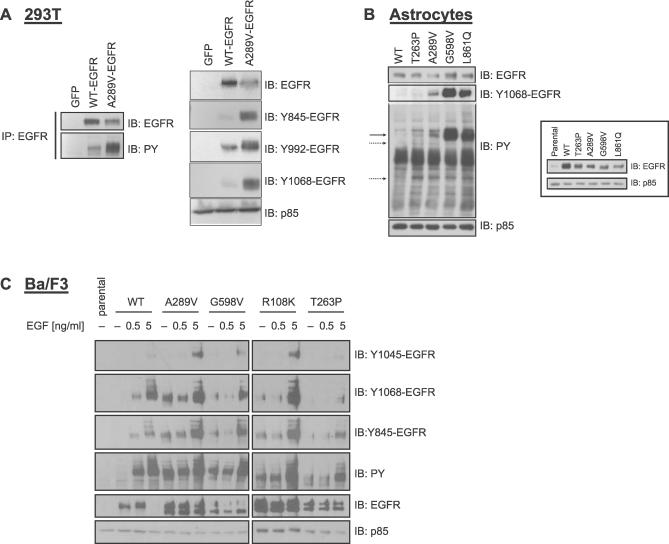
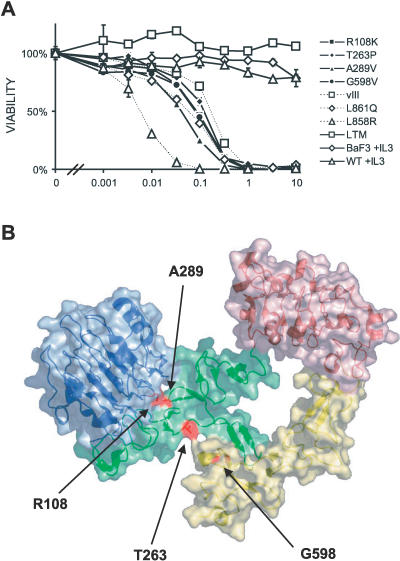
Methods and findings: Encouraged by the promising clinical activity of epidermal growth factor receptor (EGFR) kinase inhibitors in treating glioblastoma in humans, we have sequenced the complete EGFR coding sequence in glioma tumor samples and cell lines. We identified novel missense mutations in the extracellular domain of EGFR in 13.6% (18/132) of glioblastomas and 12.5% (1/8) of glioblastoma cell lines. These EGFR mutations were associated with increased EGFR gene dosage and conferred anchorage-independent growth and tumorigenicity to NIH-3T3 cells. Cells transformed by expression of these EGFR mutants were sensitive to small-molecule EGFR kinase inhibitors.
Conclusions: Our results suggest extracellular missense mutations as a novel mechanism for oncogenic EGFR activation and may help identify patients who can benefit from EGFR kinase inhibitors for treatment of glioblastoma.
Conflict of interest statement
Figures
References
-
- Hynes NE, Lane HA. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. - PubMed
-
- Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. 226–219. discussion. - PubMed
-
- Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253–1265. - PubMed
-
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
