

Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments
- PMID: 17177995
- PMCID: PMC1781470
- DOI: 10.1186/1471-2105-7-538
Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments
Abstract
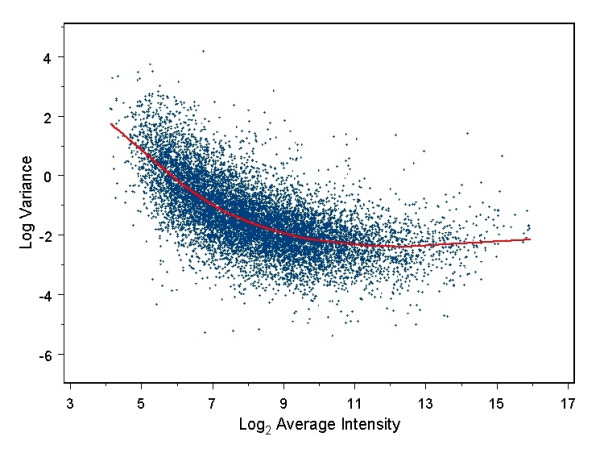
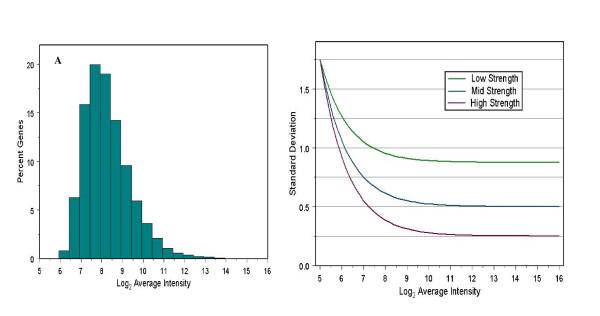
Background: The small sample sizes often used for microarray experiments result in poor estimates of variance if each gene is considered independently. Yet accurately estimating variability of gene expression measurements in microarray experiments is essential for correctly identifying differentially expressed genes. Several recently developed methods for testing differential expression of genes utilize hierarchical Bayesian models to "pool" information from multiple genes. We have developed a statistical testing procedure that further improves upon current methods by incorporating the well-documented relationship between the absolute gene expression level and the variance of gene expression measurements into the general empirical Bayes framework.
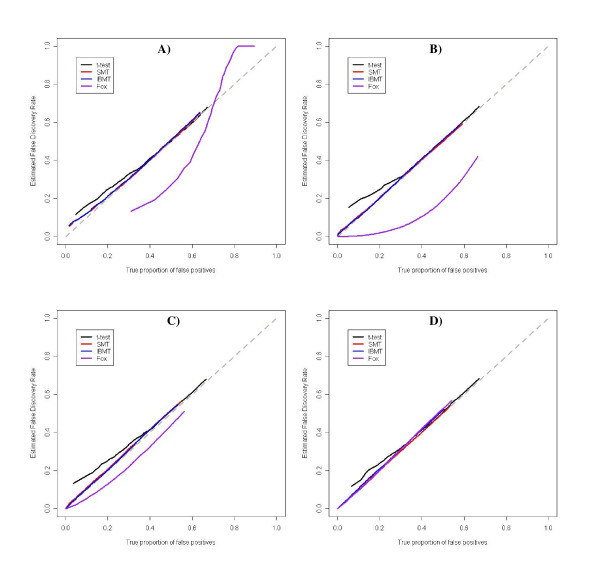
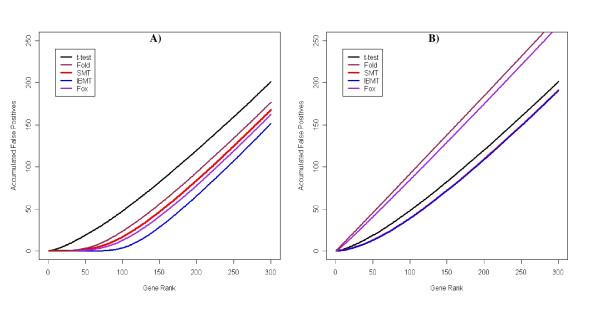
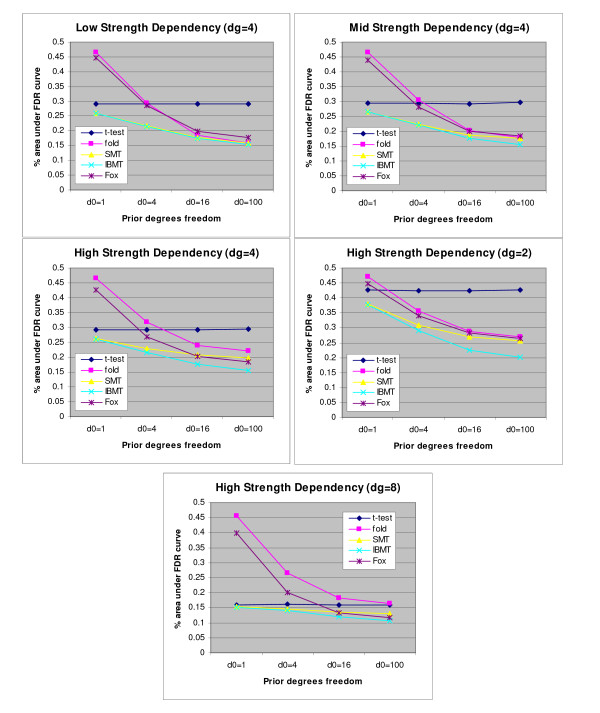
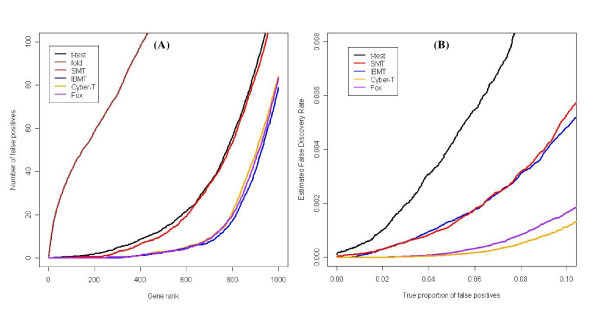
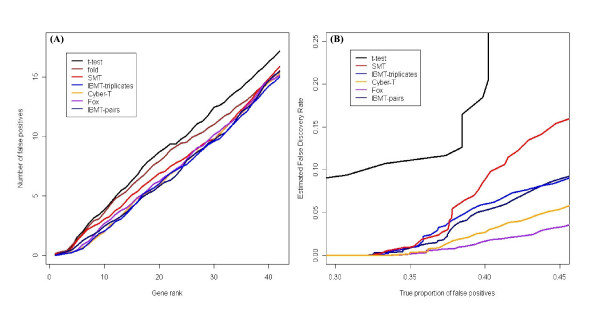
Results: We present a novel Bayesian moderated-T, which we show to perform favorably in simulations, with two real, dual-channel microarray experiments and in two controlled single-channel experiments. In simulations, the new method achieved greater power while correctly estimating the true proportion of false positives, and in the analysis of two publicly-available "spike-in" experiments, the new method performed favorably compared to all tested alternatives. We also applied our method to two experimental datasets and discuss the additional biological insights as revealed by our method in contrast to the others. The R-source code for implementing our algorithm is freely available at http://eh3.uc.edu/ibmt.
Conclusion: We use a Bayesian hierarchical normal model to define a novel Intensity-Based Moderated T-statistic (IBMT). The method is completely data-dependent using empirical Bayes philosophy to estimate hyperparameters, and thus does not require specification of any free parameters. IBMT has the strength of balancing two important factors in the analysis of microarray data: the degree of independence of variances relative to the degree of identity (i.e. t-tests vs. equal variance assumption), and the relationship between variance and signal intensity. When this variance-intensity relationship is weak or does not exist, IBMT reduces to a previously described moderated t-statistic. Furthermore, our method may be directly applied to any array platform and experimental design. Together, these properties show IBMT to be a valuable option in the analysis of virtually any microarray experiment.
Figures
References
-
- Efron B, Tibshirani R, JD S, Tusher V. Empirical bayes analysis of a microarray experiment. J Amer Stat Assoc. 2001;96:1151–1160. doi: 10.1198/016214501753382129. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
