

Evidence for large inversion polymorphisms in the human genome from HapMap data
- PMID: 17185644
- PMCID: PMC1781354
- DOI: 10.1101/gr.5774507
Evidence for large inversion polymorphisms in the human genome from HapMap data
Abstract
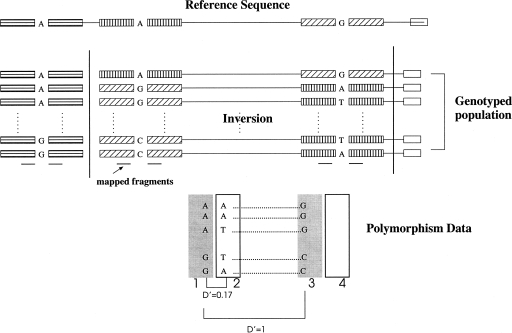
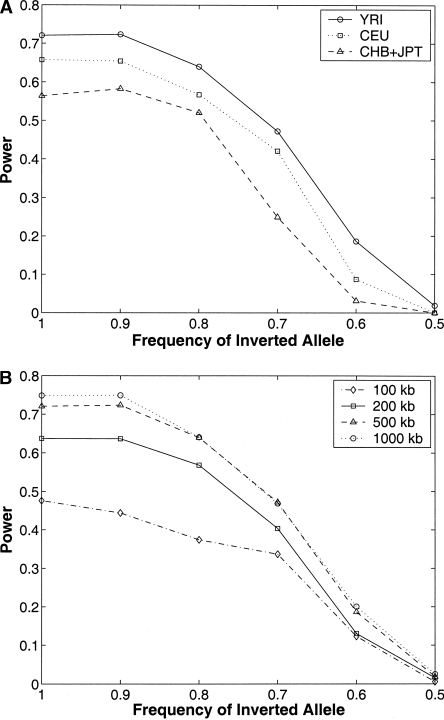
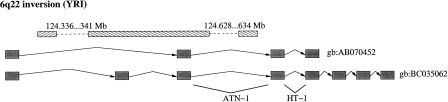
Knowledge about structural variation in the human genome has grown tremendously in the past few years. However, inversions represent a class of structural variation that remains difficult to detect. We present a statistical method to identify large inversion polymorphisms using unusual Linkage Disequilibrium (LD) patterns from high-density SNP data. The method is designed to detect chromosomal segments that are inverted (in a majority of the chromosomes) in a population with respect to the reference human genome sequence. We demonstrate the power of this method to detect such inversion polymorphisms through simulations done using the HapMap data. Application of this method to the data from the first phase of the International HapMap project resulted in 176 candidate inversions ranging from 200 kb to several megabases in length. Our predicted inversions include an 800-kb polymorphic inversion at 7p22, a 1.1-Mb inversion at 16p12, and a novel 1.2-Mb inversion on chromosome 10 that is supported by the presence of two discordant fosmids. Analysis of the genomic sequence around inversion breakpoints showed that 11 predicted inversions are flanked by pairs of highly homologous repeats in the inverted orientation. In addition, for three candidate inversions, the inverted orientation is represented in the Celera genome assembly. Although the power of our method to detect inversions is restricted because of inherently noisy LD patterns in population data, inversions predicted by our method represent strong candidates for experimental validation and analysis.
Figures





References
-
- Andolfatto P., Depaulis F., Navarro A., Depaulis F., Navarro A., Navarro A. Inversion polymorphisms and nucleotide variability in Drosophila. Genet. Res. 2001;77:1–8. - PubMed
-
- Bagnall R.D., Waseem N., Green P.M., Giannelli F., Waseem N., Green P.M., Giannelli F., Green P.M., Giannelli F., Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168–174. - PubMed
-
- Conrad D., Andrews T., Carter N., Hurles M., Pritchard J., Andrews T., Carter N., Hurles M., Pritchard J., Carter N., Hurles M., Pritchard J., Hurles M., Pritchard J., Pritchard J. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. - PubMed
-
- Crawford D., Bhangale T., Li N., Hellenthal G., Rieder M., Nickerson D., Stephens M., Bhangale T., Li N., Hellenthal G., Rieder M., Nickerson D., Stephens M., Li N., Hellenthal G., Rieder M., Nickerson D., Stephens M., Hellenthal G., Rieder M., Nickerson D., Stephens M., Rieder M., Nickerson D., Stephens M., Nickerson D., Stephens M., Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 2004;36:700–706. - PubMed
-
- Deutz-Terlouw P.P., Losekoot M., Olmer R., Pieneman W.C., de Vries-v d Weerd S., Briët E., Bakker E., Losekoot M., Olmer R., Pieneman W.C., de Vries-v d Weerd S., Briët E., Bakker E., Olmer R., Pieneman W.C., de Vries-v d Weerd S., Briët E., Bakker E., Pieneman W.C., de Vries-v d Weerd S., Briët E., Bakker E., de Vries-v d Weerd S., Briët E., Bakker E., Briët E., Bakker E., Bakker E. Inversions in the factor VIII gene: Improvement of carrier detection and prenatal diagnosis in Dutch haemophilia A families. J. Med. Genet. 1995;32:296–300. - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous